Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2021)Volume 10, Issue 4
Aqueous solubility and gastrointestinal permeability are the key determinant of drug bioavailability. The aim of this study was to improve solubility and dissolution of Meloxicam (MLX) by solid dispersion using Ziziphus spina-christi Gums (ZSCG) as drug carrier. A 32 full factorial design was used to study the effect of selected independent variables (drug-carrier ratio and kneading time) on the quality of prepared solid dispersions and to identify the optimized formula. Compatibility between MLX and carrier was proved by Fourier Transform Infrared Spectroscopy (FTIS) and the optimized formula was compressed into tablets. Results revealed that all prepared solid dispersions showed an increase in solubility over pure MLX with 10 folds increase in solubility obtained in F7. Both formulation factors exerted a significant effect (p value less than 0.05) on solubility and practical percentage yield. Formulated tablets fulfilled all compendial specifications for quality control. Dissolution profile of formulated tablets was better than a commercial brand of MLX tablets in terms of mean dissolution time which was found to be 8.35 minutes and dissolution efficiency in 30 minutes (43.56%) for the formulated tablets. Analysis of the dissolution data indicated the best fitting with Weibull and first-order with R2 of 0.9850 and 0.9813 respectively, proving that they were immediate release tablets. In conclusion, solubility and dissolution rate of MLX was enhanced by preparing its solid dispersion using ZSCG. These results are promising for more solubility enhancement upon further characterization and modification of the extracted gums.
Meloxicam; Ziziphus Spina-Christi Gums (ZSCG); Solid dispersion; Kneading; Dissolution rate; Solubility
Oral drug delivery is the simplest, most convenient and desirable way of administering therapeutic medicinal agents in particular for solid dosage forms; because of its greater stability, smaller bulk, accurate dosing and ease of manufacturing [1-3]. For any orally administered drug product the principal parameters that control the rate and extent of drug absorption are its aqueous solubility and gastrointestinal permeability with aqueous solubility being the most important property for developing a formulation [4-6]. Formulation development of such drugs would prove to be failed for oral delivery as the low solubility in aqueous gastrointestinal fluid together with lower dissolution rate lead to poor bioavailability, therefore, enhancement of aqueous solubility and dissolution rate of poorly soluble drugs remains one of the biggest challenges during drug formulation development [7,8]. Various physical or chemical approaches are available in order to improve the solubility of poorly soluble drugs, of them is solid dispersion technique which remains one of the main promising methods for improving solubility because of its simple preparation, ease of optimization, effectiveness and reproducibility [9,10]. Dispersion of drug in carriers (eutectic mixtures, solid solutions, solid dispersions) is one of the most commonly employed strategies to improve the solubility and oral bioavailability of poorly water-soluble drugs. Various hydrophilic carriers such as Poly-Ethylene-Glycol (PEG), Poly-Vinyl-Pyrrolidone (PVP), hydroxypropyl cellulose, hydroxypropylmethyl cellulose, gums, sugar mannitol, urea, hydroxypropylmethyl cellulose phthalate, gelucires, eudragits and chitosan have been investigated for improvement of dissolution characteristics and bioavailability of poorly aqueous soluble drugs [11-14].
Research for alternative natural carriers such as guar gum, xanthan gum, hupu gum, and locust bean gum has been increasing to suit for the industrial applications as well as to reduce the production cost and toxic effects. Recently, many natural polymers have been evaluated for their use in new applications [15]. Many researches have been conducted using natural carriers for developing solid dispersion in order to enhance the solubility of poorly soluble drugs. Shah et al., studied the effect of modified guar gum on the dissolution profile of Licofelone and results showed greater increase in the dissolution rate [16]. Sharma et al., applied carboxymethylcellulose sodium and xanthan gum for the formulation of clopidogrel bisulphate solid dispersion by kneading method and results showed a comparable dissolution rate enhancement for the two carriers [17].
MLX is a potent Non-Steroidal Anti-Inflammatory Drug (NSAID) and a selective cyclooxyginase-2 COX 2 inhibitor [18]. So it is a potent anti-inflammatory analgesic agent with a more favorable gastrointestinal safety profile than non-selective NSAIDs [19,20]. It is indicated for the treatment of rheumatoid arthritis, osteoarthritis, and other joint diseases. It falls under the BCS class II, poorly soluble and highly permeable drug so it has dissolution rate limited drug absorption, and this gives rise to formulation difficulties during its development for oral delivery [21]. Literature survey revealed that previous researchers had selected solid dispersion technique for enhancing solubility of MLX. Some used poloxamer 188, PEG 6000, others tried β-cyclodextrin alone or with PVP K-30 and sodium lauryl sulphate PEG 4000, mannitol and hydroxyethyl cellulose were used by Pathak [22-28]. Recently, Vinod successfully enhanced the solubility and dissolution of MLX by using sodium citrate [29]. The ability of MLX to form intermolecular interaction with various hydrophilic carrier was due to the fact that MLX can be protonated at the thiazolic nitrogen atom and deprotonated at hydroxyl or secondary amine groups forming a physical bonding in form of hydrogen bond for example with various hydrophilic carriers [30] (Figure 1).

Figure 1: Chemical structure of MLX.
To our knowledge, no previous studies have been conducted on the use of the gums extracted from Ziziphus spina-christi as drug carrier for solid dispersion; so, the aim of this study was to improve the solubility and the dissolution rate of MLX using solid dispersion technique by employing different ratios of ZSCG.
Plant materials, Ziziphus Spina-Christi (ZSC) fruits were obtained from Wad Madani market and authenticated by Department of Pharmacognosy, Faculty of Pharmacy, and University of Gezira. Meloxicam was obtained as a gift from Azal Pharmaceutical Company (Khartoum, Sudan). Micro-Crystalline Cellulose 102 (MCC 102) and cross carmellose sodium were obtained as a gift from Humavit Drugs International Co. Ltd (Khartoum, Sudan). Lactose monohydrate, talc and magnesium stearate were obtained as a gift from Amipharma Laboratories Ltd. (Khartoum, Sudan).
Methanol was obtained from LOBA CHEME Pvt. Ltd (India). Concentrated hydrochloric acid was obtained from Atom scientific (UK). Acetone, ethanol and di-potassium hydrogen orthophosphate were obtained from SD-Fine Chem. Ltd., (India). Potassium di-hydrogen orthophosphate was obtained from Central Drug House (P) Ltd., (India).
Extraction of gums from ZSC fruits
ZSC fruits were washed thoroughly and soaked in an excess of distilled water for 72 h in arefrigerator. Fruit bulb was then manually separated and allowed to stand for 24 h in a refrigerator. The supernatant was taken by decantation as a clear mucilaginous solution, concentrated to one third of its volume to which acetone was added in excess (1:2) and stirred to precipitate the polysaccharides. The precipitate was then freeze dried using freeze drier (AMSCO/FINN-AQUA, Germany) and triturated as faint brown fine powder. The obtained powder was kept in dry amber screw-capped glass container until further use [31,32].
Preparation of solid dispersion
Physical mixtures of MLX and ZSCG were prepared by mixing accurate weight of MLX with extract in three different drug: polymer; 1:0.5, 1:1 and 1:2 for 5 min using glass mortar and pestle. The physical mixture was triturated using a small volume of ethanol-water (1:1) solution to give a thick paste, which was kneaded at three kneading times; 10, 20 and 30 minutes thereafter, the obtained paste was dried at 45°C in an oven (Nuve®, Turkey). The dried mass was pulverized, passed through 60 mesh sieve size, and then weighed, transferred to an amber colored, airtight container, stored in a desiccator at 30 ± 1°C [23].
Evaluation of solid dispersion
Determination of the percentage yield: The percentage yield is a useful parameter which evaluates the efficiency of solid dispersion preparation technique. The percentage yield was calculated using the following equation:
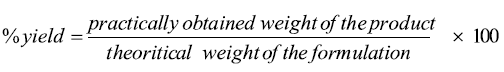
Drug content determination: Sample equivalent to 10 mg of the MLX solid dispersion was dispersed in 10 ml of methanol. The suspension formed was filtered using membrane filter paper, suitably diluted with methanol and spectrophotometrically assayed for drug content at 375 nm using 7315 UV/VIS spectrophotometer, (Jenway®, England). The drug content was calculated from the calibration curve constructed at concentration range between 5 and 30 μg/ml [33].
Factorial design: A 32 full factorial design (Design Expert version 7) was used to systematically study the influence of the individual and combined effect of independent selected variables namely MLX: ZSCG (X1) and the kneading time (X2) on the dependent variables which were the solubility in water and the practical yield percent, and thereafter determining the optimized MLX solid dispersion formula according to the obtained experimental results which would be compressed into tablets.
Fourier Transform Infrared Spectroscopy (FTIR)
The IR spectra of pure MLX, ZSCG and optimized solid dispersion formula were recorded on FTIR spectrophotometer using Shimadzu IR Tracer 100 (Kyoto®, Japan) to exclude any possible interaction between the drug and the carrier. Samples of 2–3 mg were mixed with about 400 mg of dry potassium bromide then compressed into transparent disks under pressure of 10.000-15.00 psi. The IR spectra were recorded at scanning range from 4000 -500 cm-1 and resolution of 4 cm-1 [34].
Formulation of MLX solid dispersion tablets
Tablets containing solid dispersion equivalent to 7.5 mg MLX were prepared from the optimized solid dispersion by direct compression method. Lactose monohydrate was used as diluent to adjust the weight of the tablet to 220 mg. Microcrystalline cellulose, crosscarmellose sodium, talc and magnesium stearate were used as binder, disintegrant, glidant and lubricant respectively. All formulated tablets were evaluated physically in term of hardness, friability, thickness, weight variation and disintegration as per official method [35].
In vitro dissolution studies
All dissolution studies of the prepared tablets were performed using USP apparatus 2 RC-6 Dissolution tester (Gouming®, China), with paddle stirrer manual-sampling dissolution bath, at 75 ± 1 rpm in 900 ml phosphate buffer adjusted to pH=7.5 using pH-meter (Jenway®, England), as dissolution medium.10 ml sample aliquots were withdrawn at 5, 10, 20, 30, 45 and 60 min using 10 ml syringe, with medium replacement. All samples were filtered through 0.45 μm membrane filter and the amount of drug dissolved was determined spectrophotometrically using 7315 UV– VIS spectrophotometer at 375 nm [36].
Dissolution profile of MLX solid dispersion tablets compared to a marketed brand of MLX
Model-dependent approach: In vitro drug release data were fitted to various release kinetic models including; zero-order, first-order, Higuchi, Hixson-Crowell cube root, Korsemeyer–Peppas, and Weibull model employing the following set of equations in order to verify the release kinetics of the drug as various qualitative and quantitative changes in a formulation altered drug release and in vivo performance:
Zero-order model: M0 — Mt = k0t
First-order model: Ln(M0/Mt) = K1t
Higuchi model: 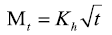
Hixson–Crowell cube root model: 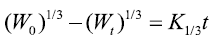
Korsemeyer–Peppas model: 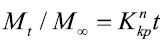
Weibull model: 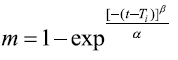
Where M0, Mt, and M∞ correspond to the drug amount taken at time equal to zero, dissolved at a particular time, t, and at infinite time, respectively. The terms W0 and Wt refer to the weight of the drug taken initially and at time t, respectively. Various other terms; k0, k1, Kh, k1/3, and Kkp refer to the release kinetic constants obtained from the linear curves of zero-order, first-order, Higuchi, Hixson–Crowell cube root, and Korsemeyer–Peppas model, respectively and for Weibull model, m represents the drug fraction accumulated in the solution on time t, α defines the timescale of the process, the parameter (Ti) represents the latency time of the release process, many times being zero and the form parameter (β) characterizes the type of curve [23].
Model-independent approach
Dissolution efficiency at 30 min. (DE30): For each sample, the percentage dissolution efficiency at 30 min. was calculated as the percentage ratio of the area under the dissolution curve up to 30 min. to that of the area of the rectangle described by 100% dissolution at the same time point, and is defined as follows:
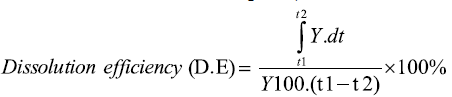
Where y is the percentage of dissolved product. D.E. is then the area under the dissolution curve between time points t1 and t2 expressed as a percentage of the curve at maximum dissolution, y100, over the same time period [37].
Mean Dissolution Time (MDT): The mean dissolution time was used to calculate the in vitro/in vivo correlation of dissolution profiles, to model the input function of the drug absorption to test the equivalence of two dissolution profiles and to compare different profiles statistically [38,39].
Mean dissolution time is determined from the accumulative curves of dissolved MLX as function of time [40].
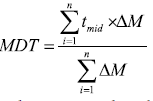
Where i, is the dissolution sample number, n is the number of dissolution times, tmid is the time at mid-point between times ti and ti-1, ΔM is the amount of MLX in μg dissolved between times ti and ti-1.
Fit factors: Fit factors, namely, the difference factor f1, and the similarity factor f2 contrast the difference between the percent of drug dissolved per unit time of a test with that of a reference formulation. F2 is more sensitive in highlighting the difference between two dissolution profiles so it was adapted here as a comparative parameter to compare the dissolution profile of the formulated MLX solid dispersion tablets and a reference marketed brand of MLX. This was calculated by the software DD solver programme [41].
Extraction of the gums
Gums from ZSC fruit bulb were obtained by acetone precipitation technique. The extract was found to be pale brown easily flowing powder that can be easily ground into different particle sizes. Powder passed through sieve no.80 was collected.
Each 1 kilogram of a whole ZSC fruit produced 5 gram of the extract indicating a percentage yield of 0.5%.
Characterization of the solid dispersions
MLX solid dispersion was prepared using different ratios of MLX to ZSCG and kneading times and the results of the characterization in term of percentage yield, drug content and solubility were illustrated in the percentage yield for all solid dispersions was found to be between 83.4-95.7%. While the drug content was found to be within the pharmacoepial specifications (85-115%) in the range of 98.2-102.56%. Regarding the solubility, all prepared solid dispersion showed enhanced aqueous solubility over pure MLX. Maximum increase in solubility was achieved in F9 prepared using MLX to ZSCG ratio of 1:2 with kneading time 30 min. where the solubility was 168.58 ± 0.0018 μg/ml with ten folds increase in solubility compared to pure MLX which was found to be 16.6 ± 0.0029 μg/ml (Table 1).
| Formula code | Variable level | Response | |||
|---|---|---|---|---|---|
| MLX:ZSCG | Kneading | % yield | % content | Solubility (µg/ml) | |
| time | |||||
| Pure MLX | 0 | - | - | - | 16.66 ± 0.0029 |
| F1 | -1 | -1 | 87.8 | 100.5 | 78.21 ± 0.0004 |
| F2 | -1 | 0 | 86.8 | 98.2 | 99.37 ± 0.0004 |
| F3 | -1 | 1 | 83.4 | 100.25 | 107.71 ± 0.0030 |
| F4 | 0 | -1 | 94.2 | 99.35 | 112.17 ± 0.0012 |
| F5 | 0 | 0 | 90.9 | 102.3 | 118.36 ± 0.0018 |
| F6 | 0 | 1 | 85.5 | 102.56 | 124.56 ± 0.0021 |
| F7 | 1 | -1 | 95.7 | 100.25 | 155.54 ± 0.0004 |
| F8 | 1 | 0 | 94.9 | 99.49 | 160.46 ± 0.0012 |
| F9 | 1 | 1 | 93.5 | 99.74 | 168.58 ± 0.0018 |
| Coded item | |||||
| -1 | 01:00.5 | 10 | |||
| 0 | 1:01 | 20 | |||
| 1 | 1:02 | 30 | |||
Table 1: Formulation factors and physicochemical characteristics of MLX-ZSCG based solid dispersions.
Factorial design
As shown in Table 2, the ratio of MLX to the ZSCG in solid dispersion had a significant positive effect on both dependent variables (solubility and % yield) with P value of 0.0003 and 0.0081 for solubility and percentage yield respectively. Whereas, kneading time had a significant positive effect on the solubility with P value of 0.0355 and significant negative impact on the percentage yield with P value of 0.0473 as illustrated in Table 3.
| Source | Sum of | df | Mean | F value | P-value |
|---|---|---|---|---|---|
| squares | square | ||||
| Model | 7325.37 | 4 | 1831.34 | 62.09 | 0.0007 |
| A: Drug: Carrier | 6817.36 | 2 | 3408.68 | 115.56 | 0.0003 |
| B: Kneading | 508.01 | 2 | 254.01 | 8.61 | 0.0355 |
| time | |||||
| Residual | 117.99 | 4 | 29.5 | ||
| Cor total | 7443.36 | 8 |
Table 2: Effect of MLX-ZSCP ratio and kneading time on solubility (ANOVA).
| Source | Sum of squares | df | Mean square | F | P-value |
|---|---|---|---|---|---|
| value | |||||
| Model | 154.04 | 4 | 38.51 | 13.7 | 0.0132 |
| A: drug: carrier | 113.58 | 2 | 56.79 | 20.21 | 0.0081 |
| B: Kneading | 40.46 | 2 | 20.23 | 7.2 | 0.0473 |
| time | |||||
| Residual | 11.24 | 4 | 2.81 | ||
| Cor total | 165.28 | 8 |
Table 3: Effect of MLX-ZSCP ratio and kneading time on practical percentage yield (ANOVA).
Fourier Transform Infrared Spectroscopy (FTIR)
Figure 2 shows the spectra of MLX, ZSCG, and optimized formula of SD. The spectrum of MLX characteristic peaks at 3,288.63 cm-1 (N-H stretching vibrations), 1,620.21 cm-1 (C=N stretching vibrations), and 1,157.29 cm-1 (S=O stretching vibrations), representing the main functional groups present in MLX structure.

Figure 2: Fourier transform infrared spectra of (a) MLX, (b) ZSCG and (c) Optimized solid dispersion of MLX.
Meanwhile, the spectrum of the ZSCG as appears in the characterized by a broad stretching intense characteristic peak near 3290.56 cm-1 which would be due to hydroxyl stretching vibration of the polysaccharide fractions, and a weak C–H stretching vibration band in the region of 2927.94 cm-1. The band towards 1743.65 25 cm-1 was attributed to stretching vibration of C=O in the protonated carboxylic acid, which resulted from the presence of uronic acids in the gums.
The spectrum of MLX in the optimized formula of solid dispersion was almost unchanged. However, the spectrum of solid dispersion exhibited a decrease in the intensity of N-H stretching vibrations and C=N stretching vibrations (Figures 2a-2c).
Postcompression evaluation of the formulated MLX solid dispersion tablets
All compressed tablets of MLX appeared glossy and shiny with an even surface free from pitting, sticking, lamination and any other tableting defects. They were all complied with USP limits set for hardness (4.17 ± 1), friability (0.83% loss), weight variation (220.635 ± 4.309) and disintegration (2.75 min).
Dissolution studies for MLX solid dispersion tablets
Dissolution studies were carried out for the optimized solid dispersion tablets according to the USP specifications using phosphate buffer pH 7.5. All tablets tested complied with the USP specifications for the dissolution which indicates that no less than 70% (Q+5%) of the API should be dissolved in 30 minutes for the immediate release tablets (Figure 3) illustrates the mean percent dissolved at each time point with the relative standard deviation (RSD). The MLX released at 5 min. was 63.44 ± 10.41%, and 94.98 ± 2.04% at 30 min. At 60 min. all amount of MLX presents in the tablets was released into the dissolution medium.

Figure 3: The dissolution profiles of the optimized MLX solid dispersion tablets and the reference marketed brand.
Dissolution profile comparison between formulated tablets and marketed brand of MLX
The optimized MLX solid dispersion tablets and the reference brand showed a comparable dissolution profile. Both model-dependent and model-independent approaches had been adopted for comparison.
Model-dependent approach: The dissolution profiles corresponding to MLX solid dispersion tablets and the marketed brand of MLX were evaluated be fitting the experimental data to different drug release kinetic models. The Weibull model provided the best adjustment curve for both the formulated solid dispersion tablets and the marketed brand of MLX with determination coefficient (R2) of 0.9850 and 0.9982 respectively, and β value ˂1 explaining that the shape of the curve was parabolic, displaying high initial slope and a consistent exponential character, followed by the first-order model which also represent good fitting with R2 of 0.9813 and 0.9963 for test tablets and reference brand respectively (Table 4).
| Model | Statistics | MLX solid dispersion tablets | Marketed brand |
|---|---|---|---|
| Zero-order | R2 | -0.3784 | 0.1335 |
| K0 | 2.374 | 2.268 | |
| First-order | R2 | 0.9813 | 0.9963 |
| K1 | 0.213 | 0.115 | |
| Higuchi | R2 | 0.5956 | 0.8335 |
| KH | 16.76 | 15.716 | |
| Korsmeyer-Peppas | R2 | 0.7013 | 0.8687 |
| Kkp | 57.179 | 35.877 | |
| n | 0.153 | 0.268 | |
| Hixson-Crowel | R2 | -1.446 | 0.9458 |
| KHC | 0.026 | 0.024 | |
| Weibull | R2 | 0.985 | 0.9982 |
| Td | 4.951 | 8.376 | |
| β | 0.316 | 0.782 |
Table 4: Dissolution profiles of the optimized MLX solid dispersion tablets compared to a marketed brand using different mathematical drug release models.
Model-independent approach: The determination of dissolution efficiency and mean dissolution time values are useful methods to reduce each curve to a single number. The dissolution efficiency in 30 minutes (DE30%) was found to be 43.56% and 41.62% for the test and reference brand of MLX respectively, while the mean dissolution time (MDT) was found to be 8.35 minutes for the formulated tablets and 9.9 minutes for the reference brand.
Fit factors namely; similarity (F2) and difference factors (F1) have been accepted by FDA Centre for Drug Evaluation and Research (CDER) as a rating criterion of similarity between two in vitro dissolution profiles [42]. This parameter was also calculated for the test product in order to assess its bio-equivalency to the reference brand. According to the guidelines which state that, F2 values greater than 50 should ensure equivalence between the dissolution curves, indicating an average difference of no more than 10% at the sample time points. So that the dissolution curves corresponding to the test product and the reference brand would be dissimilar since the value of F2 was found to be 47.16.
The present work aimed to enhance the dissolution characteristics of MLX through formulating it in the form of solid dispersion by kneading method. Drug content, solubility and dissolution rate determination were done using calibration curve of MLX. Relationship between concentration and absorbance was linear with R2=0.9993.
Drug carrier and kneading time were studied regarding their effect on the solubility and practical percentage yield of the prepared MLX solid dispersion. Increase in solubility was observed in all formulae of the prepared solid dispersion compared to pure MLX (Table 1). Possible mechanisms for solubility enhancement of solid dispersion has been given by Chiou and Riegelman including; particle size reduction, solubilization effect resulting from the high concentrations of the carrier in the diffusion layer surrounding the solid dispersion, reduced aggregation of drug particles and improvement of wettability [43] (Figure 4a).

Figure 4: Effect of MLX: ZSCG and kneading time on (a) Practical Yield percent, (b) Solubility.
As the concentration of the carrier was increased, the solubility was increased significantly, this can be justified by the theory introduced by Corrigan in 1985 which stated that the dissolution rate of the polymer alone was shown to be equivalent to the release rate of the drug from a drug-carrier system with a high carrier fraction [44]. This finding was confirmed by Dubois and Ford in 1985 who investigated the release rates of various drugs from polyethylene glycol (PEG) 6000 systems. If a solid dispersion was formed by the drug and the carrier, the rate of dissolution was determined to be equivalent for all investigated drug-carrier systems regardless of the drug properties [45]. Also this finding was in an agreement with that of Ghareeb et al., Al-nima et al. and Umesh et al. who found that the solubility of MLX was increased with increasing polymer concentration in solid dispersion formula [21-23]. Percentage yield as seen in Figure 4b was also affected significantly by carrier concentration in such way that as carrier concentration was increased the percentage yield was increased. However this finding was in contrast of that of Ghareeb et al. who found that yield percent decrease at higher polymer ratio, and this was attributed to difficulty of sieving when higher polymer ratio was used [23].
Kneading time had less positive effect on solubility than the drug: carrier, as with increasing kneading time the solubility was increased due to better dipersibility of drug particles within the soluble carrier. Also the percent yield was significantly affected positively by kneading time this may be due to less viscosity of the solid dispersion formed with the carrier making scrapping and sieving it easy and with minimum loss.
The Fourier Transform Infrared (FTIR) represents an efficient method to exclude any interaction that may occur between the drug and carrier in solid dispersion and is the one of the most popular tool for characterization of solid dispersion.
The spectrum of solid dispersion exhibited a decrease in the intensity of N-H stretching vibrations and C=N stretching vibrations. These changes in the intensities of the spectrum of the solid dispersion may suggest the physical interaction through hydrophobic bonds such as hydrogen bonding between the drug and the polysaccharide used. The spectrum of solid dispersion exhibited no significant changes in the location or width of the characteristic infrared peaks of MLX, which indicated that there was no chemical interaction between MLX and the carrier.
Dissolution studies for the formulated tablets was carried out according to USP specifications using phosphate buffer pH 7.5 as MLX is an acidic drug (pKa, 1.1), practically insoluble in water at physiological pH (solubility is 12 μg/mL) and has a zwitterionic property with two pKa values (pKa1=1.09, pKa2=4.18) [46,47]. The percentage of ionized drug and the solubility increase with increasing pH until the highest solubility reported is reached in phosphate buffer pH 10, decreasing pH leads to an increase in the ratio of non-ionized to ionized drug combined with a decrease in solubility [46]. Previous pharmacokinetics studies have shown that MLX has prolonged absorption with Tmax of longer than 5h, indicating the slow absorption of meloxicam after oral administration, so that a dissolution media adopted was phosphate buffer pH 7.5 [48,49]. Formulated MLX solid dispersion tablets showed a comparable dissolution profile to a marketed brand of meloxicam, since meloxicam is BCS class II, various formulators need to put in their minds the dissolution behavior of this drug because of its poor aqueous solubility with consequent poor dissolution characteristics. Additionally, for a drug to pass the pharmacopeial dissolution test, not less than 70% of stated amount must be dissolved in 30 min after the administration. Because of all these reasons, various techniques were utilized during formulation of MLX for markets including for example the use of miconized powder and surfactants in order to enhance the solubility and so the dissolution behavior of the drug so as to pass the compendial requirement for the dissolution. Simionato et al. studied the dissolution profile of nine marketed brands of MLX available in Argentina and it was found that all brands release more than 70% of the stated amount during the first 30 min. of the dissolution and this prove utilization of the formulators of various techniques to improve the dissolution characteristics of poorly soluble BCS class II MLX. Regarding the kinetics of drug release, the Weibull model provided the best adjustment curve for both the formulated SD tablets and the reference brand, with the higher determination coefficients (R2) and smallest AIC values. This result was in agreement with the results of Simionato et al., who studied the release kinetics of nine brands of MLX and was concluded that the best fitting achieved was with Weibull model [36]. The first-order model also provide good fitting with R2 of 0.9813 and 0.9963 for test tablets and reference brand respectively proving that the tablets tested were immediate release and their release was dependent on the available concentration of the drug in the dissolution medium, and that the applied carrier in the formulation of solid dispersion with a drug-carrier ratio; 1:2 has no effect on the release of the drug from the tablet which indicated the absence of release retardation. This confirms that the employed carrier has a very high water solubility, enhancing only the release rate of MLX without release retarding capability. This was in accordance with study carried out by Barzegar-Jalali et al. who prepared a solid dispersion of piroxicam using microcrystalline cellulose (MCC), crosspovidone and Elaeagnus angustifolia fruit powder by cogrinding technique and concluded that drug release rate was enhanced as a consequence of increasing carrier concentration up to ratio of 1:2 [50].
The parameters mean dissolution time, MDT and dissolution efficiency at 30 min. have been used not only to describe dissolution profiles with the aim to reduce the data into a single number, but also to calculate the in vitro/in vivo correlation of dissolution profiles, to mode 1 the input function of the drug absorption to test the equivalence of two dissolution profiles and to compare different profiles statistically [38-39]. Better dissolution efficiency and less mean dissolution time for the formulated tablets when compared to the marketed brand of MLX highlighted the significance of solid dispersion technique in enhancing the dissolution behaviour of MLX.
It can be concluded that, solid dispersion is a simple and successful technique for improving the solubility and hence the dissolution behavior of MLX. ZSCG, as a drug carrier for solid dispersion, gave satisfactory result of solubility enhancement that was comparable to other conventional carriers commonly employed for formulation of solid dispersion. The results of the factorial design confirm that, formulation factors (MLX: ZSCG and kneading time) significantly influence the dependent variables; solubility and practical yield percent. Characterization studies showed that no chemical interaction was encountered between MLX and the carrier. The dissolution profile of the formulated tablets was better than that of the marketed brand of MLX in term of dissolution efficiency in 30 min. and mean dissolution time. These results encourage more characterization and/or modification of ZSCG to have more solubility enhancement.
We would like to acknowledge the staff technicians of the quality control laboratory, Faculty of Pharmacy, University of Gezira. Our thanks extended to the national pharmaceutical companies; Amipharma laboratory, Azal pharmaceutical Company and Humavit Company, Khartoum, Sudan for supplying us with required pharmaceutical materials. Also, we want to acknowledge the members of the Department of Pharmaceutics at National Ribat University, special thanks to Dr. Dawood Baraka, the head of the department.
Citation: Ismail EA, Elamin ES, Ahmed EMM, Abdelrahman M (2021) Enhancement of Aqueous Solubility of Meloxicam using Solid Dispersions Based on Ziziphus spina-christi Gums. Drug Des. 10:188.
Received: 14-Jul-2021 Accepted: 28-Jul-2021 Published: 04-Aug-2021 , DOI: 10.35248/2169-0138.21.10.e188
Copyright: © 2021 Ismail EA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.