Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Research Article - (2014) Volume 3, Issue 1
Keywords: Protease, Bacillus sp, Strain improvement, UV-induced, Ion exchange chromatography, Genetic rearrangement
Proteases are essential constituents of all forms of life on earth including prokaryotes, fungi, plants and animals. Proteases are highly exploited enzymes in food, leather, detergent, pharmaceutical, diagnostics, waste management and silver recovery [1]. Proteases [serine protease (EC. 3.4.21), cysteine (thiol) protease (EC 3.4.22), aspartic proteases (EC 3.4.23) and metalloprotease (EC 3.4.24)] constitute one of the most important groups of industrial enzymes, accounting for about 60% of the total enzyme market [2-4]. Among the various proteases, bacterial proteases are the most significant, compared with animal and fungal proteases [5] and among bacteria, Bacillus sp. are specific producers of extra-cellular proteases [6]. These enzymes have wide industrial application, including pharmaceutical industry, leather industry, and manufacture of protein hydrolysis, food industry and waste processing industry [7]. Thermo-stable proteases are advantageous in some applications because higher processing temperature can be employed, resulting in faster reaction rates, increase in the solubility of nongaseous reactants and products and reduced incidence of microbial contamination by mesophilic organisms. Proteases secreted from thermophilic bacteria are thus of particular interest and have become more useful in a range of commercial applications [8,9]. Previously Gohel et al. [10] used UV-mutagenesis and gamma mutagenesis (physical mutagen) for the strain improvement of chitinolytic enzyme producing isolate Pantoea dispersa. The isolated mutant of Pantoea dispersa showed high proteolytic enzyme activity compared to wild type strain. This area provides avenues of changes in physiological, ecological, genetical because mutation plays a key role in biological process. UV-radiation induces mutations such as frame-shift, base substitution, deletion, recombination and other type of genetic rearrangement [11]. Therefore present study deals with genetic improvement of Bacillus sp. to increase high protease production through UV-induced mutation. The results obtained from mutants were compared with wild type mutant strain had shown higher protease and stable at wide range of temperature and pH variation and resist to different inhibitors as well.
Isolation enrichment and screening of bacterial strain
Isolation and enrichment of protease producing bacteria were done by standard dilution-plate technique. For this we used soil sample collected from agricultural soil from Devsthali, Haryana, India. 5 g of soil sample added to a medium containing 0.5% (w/v) yeast extract and 1.0% (w/v) peptone for one week. Further isolation of bacteria was done on the medium mixture was composed of 0.5 g/l glucose, 0.4 g/l Na2HPO4, 0.085 g/l Na2CO3, 0.0 2 g/l ZnSO4, 0.0 2 g/l MgSO4, 0.02 g/l CaCl2 and pH was adjusted to 7.2. To solidify the medium 1.5% agar was added and the medium. A series of dilutions (10-3, 10-4 and 10-5) was prepared using sterile water. Aliquots of 0.1 ml from the different dilutions were spread on the solid growing medium. For each dilution, three replica plates were prepared. Morphologically different colonies were isolated and purified by repeated sub culturing on growing agar medium. Further Screening was carried out using milk agar plate containing; skim milk powder 0.1% and 0.2% agar further 10 μl culture filtrate were used on the surface of milk agar plate. Plates were incubated at 30°C for 48 hr to check the production of protease based on the zone formation where the filtrate was spotted. Bacteria strain was selected on the basis of ability of zone formation one efficient strain was selected and designated as RS.
UV-mutagenesis
The phenotypic mutants of Bacillus strain RS were developed by UV irradiation. Bacillus strain RS was grown overnight in above medium containing 1.0% (w/v) peptone at 37°C under shaking condition. The cells were washed twice with 0.5 M phosphate buffer (pH 8.0) and resuspended in 1 ml of the same buffer. The cells were diluted in phosphate buffer to obtain 5×106 cfu/ml and exposed to UV using a Philips 20-W germicidal lamp for 20 min, where a less than 10% survival rate was observed. The colonies were randomly selected and transferred onto skim milk agar plates (Hi-Media, India) to check the production of protease based on the zone formation. Five strain were selected based on their ability to produce highest zone formation and production of protease, further only one strain RS1 (mutant) was selected for further study. The mutants were cultured at least five times to assure incapability of reverse mutation and compared with wildtype strain RS for protease production. The effect of different pH (in buffered media) on the growth of these mutants was also studied.
Measurement of protease activity by azocoll hydrolysis
The activity of protease was assessed by azocoll hydrolysis in test tube containing 10 mg of azocoll dissolved in 10 mM potassium phosphate buffer at pH 7.5 and 0.5 ml of test sample was added. For a negative control (auto-hydrolysis), 0.5 ml of buffer was added. Tubes were vortexes for 30 seconds, following incubation for 25 min at 37ºC with agitation, tubes were placed in ice. Suspensions were filtered through what man No. 4 filter paper in glass funnels into tubes in an ice-water bath to prevent continued azocoll hydrolysis. The absorbance of the released azo-dye was measured at 520 nm against a reagent control blank. For calculation of specific protease activity, absorbance at 520 nm was multiplied by 2.965 to obtain mg azocoll hydrolyzed in 25 min at 37°C per volume of test sample [12]. One unit of protease activity was defined as the amount of enzyme that yielded an increase in absorbance at 520 nm in 25 min at 37°C. Protease activity was expressed in U/mg. Total protein concentrations were measured by the method by Lowry et al. [13].
Determination of the cellular protease activity
Cells were grown in 250 ml Erlenmayer flask containing growing medium and incubated at 30°C and 120 rpm. Cultures were harvested at early stationary phase (approximately 16 hr). The culture was centrifuged (10,000 rpm for 30 min under refrigerated condition) and the culture supernatants collected and filter sterilized by passage through a 0.2 μm pore size filter and stored in an ice-water. The cell pellet was re-suspended in an equal volume of 10 mM phosphate buffer pH 7.5 a sample of the cells in the remaining suspension was sonicated using a sonicator (SG-25) Roop Telesonic Ultrasonics Ltd. India, with variable power intensity between 3 and 7.4 W/cm2 and a frequency of 24 kHz. The sample was immersed in ice and sonicated for a total of 30 min with 1.5 min burst followed by 5 min rest on ice. After centrifugation at 10,000 rpm for 15 min, the supernatant was used to determine the protease activity and protein concentration and specific activity determined for filtered supernatant and crude extract fractions.
Ammonium sulfate precipitation and ion-exchange chromatography
Strain RS1 was grown in above medium to 45 hr. Cells was harvested and centrifuged (10,000 rpm for 30 min at 25ºC). The supernatant was then filtered using 0.45 μm pore size filters. Ammonium sulfate was poured slowly into the supernatant over a period of 10 min, allowing the salt to slowly dissolve. The supernatant was continually stirred at room temperature for an additional 25 min. Precipitates were recovered by centrifugation (10,000 rpm for 10 min at 25ºC), and dissolved in 10 mM Tris-HCl, pH 8.0. Each (NH4)2SO4 fraction was dialyzed overnight against the same buffer (1:50 volume). The protease activity and protein concentration was measured and specific activity calculated. To determine the precipitation fraction that contained the largest percent of protease activity, 20% fractions (0-100%) were retained. Protease activity and protein concentration was measured and specific activity calculated.
The dialyzed ammonium sulfate fractions were concentrated using an ultrafiltration cell (Amicon) through a membrane pore size >5,000 MW (Milipore, USA). The resulting concentrated samples (4-5 ml, 2.0 - 2.5 mg) were applied to a column (16 cm length, 2 cm diameter) of DEAE-Sepharose fast flow beads (Amersham Pharmacia, USA). The column was equilibrated with 150 ml 1M NaCl buffer pH 8.0 and washed with 150 ml of 10 mM Tris using a fast performance liquid chromatography system (Amersham Pharmacia, USA) protein were eluted with 10 mM Tris and 0 to 1.0 M NaCl gradient. Eluted fractions 4 ml were collected and absorbance was measured at 280 nm. Protein peaks were pooled then dialyzed overnight against the Tris, NaCl buffer. The protease activity and protein concentration of fractions was measured and specific activity calculated.
Characterization of protease activities
The influence of pH on proteolytic activities was determined using azocasein with citrate-phosphate buffer (0.1 M), and the influence of temperature was determined by incubating the reaction mixtures for 15 min at temperatures ranging from 30 to 90ºC in 50 mM sodium phosphate buffer (pH 7.0). 10 ml of each of the three inhibitor stock solution was added separately to the reaction mixtures, and the remaining activity was determined. Stock solution of each protease inhibitor were prepared, namely: serine protease inhibitor-100 mM phenylmethylsulfonyl-fluoride (PMSF) solution in dimethyl sulfoxide (DMSO), 100 mM 1, 10-phenanthroline solution in DMSO and 100 mg/ml pepstatin solution in DMSO.
Biochemical and molecular characterization of bacterium
The biochemical characterization of the three isolates was done in accordance with Bergey’s Manual of Systematic Bacteriology [14]. Pure culture was subjected to biochemical characterization. Gram stain was done by using gram staining kit, Capsule stain was determined with 20% copper sulfate. Catalase activity was determined based on formation of bubbles in the presence of 3% H2O2 solution. Oxidase was performed on paper discs using tetramethyl-pphenylenediamine. Nitrate reductase was detected on nitrate agar plates and DNase on DNase agar medium with methyl green as an indicator. Motility, indole formation, and urease activities were determined by using an MIUmedia kit (Hi-Media Laboratories, India). Genomic DNA extraction, amplification of 16S rRNA gene and sequence analysis was performed as described in Karn et al. [15]. The sequences were compared against the available DNA sequences in GeneBank (https://www.ncbi.nlm.nih. gov/) using the BLASTN tool. The 16S rRNA gene sequence determined in this study was deposited in the GenBank of NCBI data library under the accession number KF702315.
Biochemical characterization and identification of bacterium
The bacterium was biochemically characterized shown positive for Gram stain, motility, catalase, oxidase, and nitrate reduction test whereas negative for capsule, urease and DNAse test. Further for molecular identification 16s rRNA gene was amplified and cloned and transformed into DH5α cells. The 16s rRNA product from selected clones were then sequenced using Applied Biosystem automatic sequencer. Sequence was compared for the similarity in the GeneBank using BlastN. Pair wise alignment revealed that 16s rRNA gene of the bacterial isolate had 99% identity with Bacillus sp.
Protease activity
Bacillus strain RS1 showed clearing zones on the milk casein agar indicating the hydrolysis of the protein within the agar and thus the presence of a protease. Zones measuring 14-15 mm of complete hydrolysis of the milk casein agar occurred after plates were spotted with 10 μl cultures and incubated for 48 hr at 30°C but for wild strain RS zones were relatively smaller 10-12 mm. Further to determine the assay for measurement of protease activity was also done by azocoll assay tubes containing both the strain RS and RS1 supernatant as well as the positive control, showed hydrolysis of the dye-linked insoluble collagen. Hydrolysis caused a release of a purple dye, which allowed tubes to be immediately read by eyesight.
Growth and protease production
Absorbance was measured at every 6 hr of intervals. An RS1 and RS (wild) cell begins exponential growth approximately at 6 hr after inoculation and enters to stationary phase at approximately 48 hr. This exponential phase was observed at12-24 hr. At 48 hr the cells appeared to be in decline phase, with indication of a death phase. Protease appeared at low levels immediately after inoculation. There was a noticeable rise in the protease which begins at 12 hr during early stationary growth (Figure 1a mutant RS1, 1b wild RS). Protease activity continually increased after 18 hr through the entire growth curve measured. For the purification of protease, it was decided that culture samples should be taken at a time point later than 24 hr (early stationary) of growth (Figure 1a and 1b).
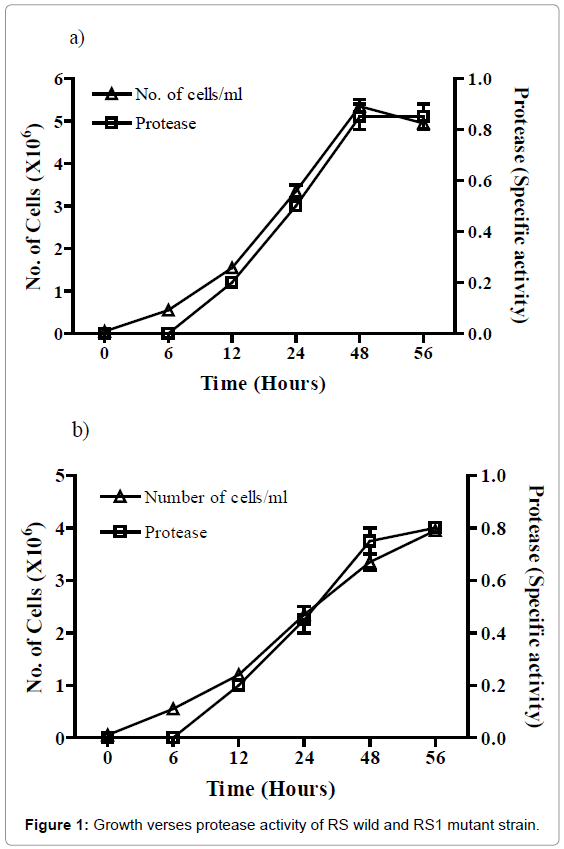
Figure 1: Growth verses protease activity of RS wild and RS1 mutant strain.
Determination of the protease
The, the filtered culture supernatant had a specific activity of 1.10, and the cells in buffer measured 0.98. The filtered culture supernatant had the highest specific protease activity and contained 86% of the overall protease percentage of the culture. The whole supernatants accounted for 86% of the protease activity, and in the crude extract (100%) in the mutant strain comparatively wild strain has little less activity shown in Table 1. This high percentage could be due to the fact that the assay was conducted at 37°C for 25 min, which allowed for cells to produce more protease. The crude extract fraction of RS had a specific protease activity (mg/mg of proteins) in culture supernatant was 0.9 and in crude extract was 0.006 whereas RS1 has 1.2 in culture supernatant and 0.009 unit in crude extract.
| Fraction recoveries | Protein (mg/ml) | Protease | Fraction recoveries | Protein (mg/ml) | Protease |
|---|---|---|---|---|---|
| Wild | Mutant | ||||
| Culture supernatant | 1.0 | 82% | Culture supernatant | 1.2 | 86% |
| Crude extract | 0.9 | 75% | Crude extract | 1.4 | 100% |
Table 1: Fraction recoveries activities of RS wild and RS1 mutant strain.
Ammonium sulfate precipitation and ion exchange chromatography
Ammonium sulfate precipitation was performed as the first step of protease purification. Ammonium sulfate fractions collected at levels of saturation of 20% (w/v) were collected and assayed for specific protease activity after dialysis. Fractions collected at 40-60% and 60- 80% ammonium sulfate contained the highest percent of specific protease activity, with a total of 57%, and accounted for 22.2% of the total protein concentration (Table 2). To more precisely define the ammonium sulfate fraction containing the highest amount of specific protease activity, smaller ammonium sulfate fractions were collected over the range of 40-80%. It was determined that the 50-70% ammonium sulfate fraction contained 48% of the protease activity (Table 2). An ammonium sulfate precipitation between 40-70% was used for purifying the protease and resulted in a 20-fold increase in specific activity compared to the un concentrated supernatant.
| Amonium sulfate | % of Total Protein concentration (Wild) | % of Enzyme activity (Wild) | % of Total Protein concentration (Mutant) | % of Enzyme activity (Mutant) |
|---|---|---|---|---|
| 0-20 | 15 | 3.7 | 19 | 4.6 |
| 20-40 | 12 | 15.2 | 14 | 17.3 |
| 40-60 | 11.2 | 36.2 | 14 | 41.1 |
| 60-80 | 11 | 21.3 | 13 | 25.3 |
| 80-100 | 13.7 | 3.24 | 16 | 4.1 |
| 50-70* | 14.2 | 48 | 17 | 55 |
Table 2: Ammonium sulfate precipitation RS wild and RS1 mutant strain.
Dialyzed ammonium sulfate precipitates (40-70%) were loaded onto a DEAE Sepharose column. Proteins were eluted using a 10 mM Tris buffer (pH 8.0) and 0 - 1.0 M NaCl at a pH of 8.0 and eluted proteins were collected, fractions were pooled, absorbance were measured, and tested for specific protease activity. This contain high protease activity (4.0 specific protease activity) found in the buffer fractions containing 0.0-0.16 M NaCl. The column purification resulted in a 44-fold increase in specific activity compared to the un concentrated supernatant, and a 2.2-fold increase compared to ammonium sulfate precipitation. The first pooled column fraction was dialyzed overnight and then reloaded onto the same DEAE-cellulose column that had been washed and re-calibrated. Protease activity was again associated with the pooled fractions at the beginning of the NaCl gradient (0.0-0.25 M NaCl), The second column purification resulted in a 49-fold increase in specific activity compared to the first column purification, increasing the first column purification by 10% of the total activity.
Characterization of proteases
The optimum temperature for the proteolytic activities of the crude enzyme extracts, prepared from RS and RS1 isolates, ranged from 40ºC to70ºC for strain RS1 and 40ºC to 60ºC for strain RS at (Figure 2a). The optimum pH for the proteolytic activities of Rs isolates was 7.5 to 8.0 for RS and 7.5 to 8.5 for RS1 (Figure 2b), However, the optimum pH of the RS1 isolate was 8.5, which was slightly higher than the pH 7.0. In the presence of PMSF proteolytic activities of all isolates were inhibited (Figure 2c), suggesting they contained serine protease. But RS1 the proteolytic activities of isolates RS and RS1were significantly inhibited by 1, 10-phenanthroline by about 47 and 50%, respectively, suggesting the presence of metalloprotease.
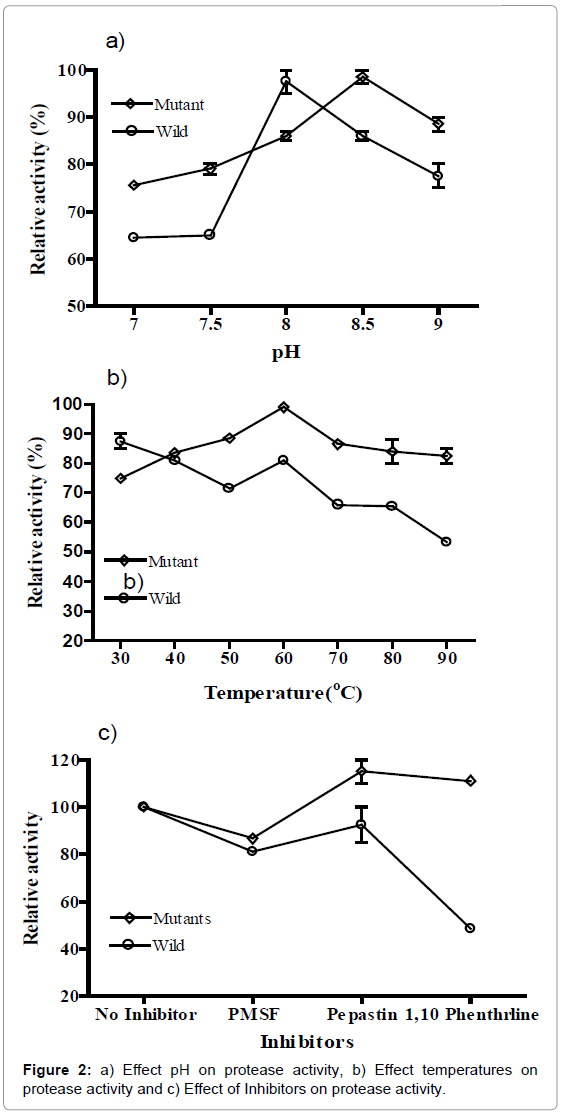
Figure 2: a) Effect pH on protease activity, b) Effect temperatures on protease activity and c) Effect of Inhibitors on protease activity.
Characterization of bacterium
The phenotypic mutants of Bacillus sp. RS1 were developed by UV irradiation and compared with the wild-type strain RS for their ability to produce protease and also observed effect of inhibitor on the production of protease. In skim milk media, RS1 has showed bigger zone as compared to RS of substrate hydrolysis. Zones of clearing were indicative of hydrolysis of the milk casein and using Azocoll as a protease substrate. Further bacterium was biochemically characterized and 16S rRNA gene sequences was also analyzed to determine the identity of bacteria isolate which showed highest similarity with the Bacillus sp. cited in the GenBank. Homology analysis of 16S rRNA gene provides suitable genetic data that can be used to determine both close and very distant relationships [16].
Production and purification of protease
In milk casein agar media, Bacillus strain RS1 showed zones of substrate hydrolysis indicating the hydrolysis of the protein within the agar and thus the presence of a protease zone was 3-4 cm lager than RS. Absorbance was continuously measured and it was increased up to 48 hr maximum protease was observed at early stationary phase. Usharani and Muthuraj [17] isolated protease from B. latrosporus had a maximum of extra cellular protease enzyme activity was at 3 days. Zambare [18] also found Bacillus sp. for protease production and maximum activity of 125.68U/ml at the end of 48 hr of incubation at 40°C. This study was conducted to investigate UV-induced Bacillus strain RS1 production of proteases and compared with wild Bacillus strain RS. Ammonium sulfate purification increased the protease activity 20-fold compared to the activity present in the unconcentrated culture supernatant. This precipitation step also decreased the overall protein concentration (70-90%) in the precipitation fraction containing the highest protease activity. Two DEAE-Sepharose column purification steps followed the ammonium sulfate purification. The protease activity was present in the 0 - 0.25 M NaCl gradient in both column runs. Ammonium sulfate precipitation followed by DEAE-Sepharose column chromatography of RS and RS1 supernatant increased specific activity 50-fold. The purification process took approximately 72 to 96 hr. Although the purification steps were conducted at room temperature and specific activity increased, because the protease was subject to auto-hydrolysis, it is likely higher yields and specific activity can be attained by preventing proteolytic activity. Increases of the RS1 protease activity using ammonium precipitation (20-fold increase) are consistent with published literature, which shows a purification (fold) range from 1.9- 96. Subsequent increases in protease activity (50-fold increase) using DEAE-column chromatography were also consistent with purification ranges from 7.8-555 [19,20].
Characterization of proteases
The optimum temperature for the proteolytic activities of the crude enzyme extracts, prepared from RS and RS1 isolates, ranged from 40ºC for strain RS1 to 70ºC for strain RS at 60ºC (Figure 2a). The optimum pH for the proteolytic activities of RS and RS1 isolates was 7.5 to 8.0 for RS and 7.5 to 8.5 for RS1 (Figure 2b), which is similar to that of other proteases produced from Bacillus sp. and B. stearothermophilus F1 [21]. Wan et al. [22] reported thermostability of protease up to 60ºC. However, the optimum pH of the RS1 isolate was 8.5, which was slightly higher than the pH 7.0 shown by the zinc proteinase of A. hydrophila [23]. Viana Daniela de et al. [24] also showed that protease activity maximum at pH 7.0. Earlier Prakasham et al. [25] reported production of protease from Bacillus sp. Neutral proteases have been previous reported in Pseudomonas sp. and Burkholderia sp. Bacillus cepacia produces a protease with an optimal pH of 6.0 [26] and a Pseudomonas fluorescens strains produce proteases active at neutral pH [27,28]. Present results indicated that RS1 protease was more active at a neutral pH but can work up to pH 8.5 with slight decrease in activity or resist pH changes and temperature. UV mutation may cause genetic change in the RS1, which may favor the bacterial strain to resist at high pH and temperature as compare to wild type. Further future study will be carried out regarding genetic changes.
Purified protease samples were assayed in the presence of inhibitors. The failure of either serine or cysteine proteinase inhibitors PMSF suggest that RS1 protease is a metalloproteinase [29]. The strongest inhibitor of protease activity was 1-10-phenanthroline, which is the preferred metalloproteinase inhibitor [29]. In the presence of PMSF proteolytic activities of of both RS and RS1 isolates were inhibited (Figure 2c). Recently Cavello et al. [30] also found that Ca2+ enhances protease activity and PMSF strongly inhibit the protease activity suggesting presence of thiol-dependent serine protease. But proteolytic activities of isolates RS and RS1were significantly inhibited by 1, 10-phenanthroline by about 47 and 50%, respectively, suggesting the presence of metalloprotease.
The authors are thankful to Biotechnology Engineering Department, Ambala College of Engineering and Applied Research for providing the facilities for the work.