Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Review Article - (2014) Volume 3, Issue 2
Nearly all cancers are linked by the inexorable phenotype of metastasis as malignant growths have the capability to spread from their place of origin to distant sites throughout the body. While different cancers may have various propensities to migrate towards specific locations, they are all linked by this unifying principal. Unlike most neoplasms, leukemia has inherent cell motility as leukocytes are required to move throughout the vascular system, suggesting that no mutations are required for anchorage independent growth. As such, it seems likely that leukemias are inherently metastatic, endowed with cancer’s deadliest phenotype simply due to cell of origin. This article presents the biology of metastasis development and how leukemia cells are inherently provided this devastating phenotype. It is then elucidated how clinicians may be able to exploit the motility of leukemia and metastatic emboli of other cancer types through an approach known as Sonodynamic Therapy (SDT), a treatment modality that combines chemotherapeutic agents with ultrasonic irradiation to preferentially damage malignant cells. As experimental evidence has indicated, SDT is a promising therapeutic approach in need of clinical testing for further validation.
<The heterogeneity and extent of diversity among known cancer types is truly immense. Even within a given cancer type, there are various histologies, aberrant biochemical pathways and mutations that permeate a truly unique disease for an individual patient. However, all cancers are linked by the inexorable phenotype of metastasis. In other words, each cancer has the capability to spread from its place of origin to distant sites throughout the body. While such cancers may have various propensities to migrate towards specific locations, they are all linked by this unifying principal. In fact, more than 90% of patient mortality due to cancer is a direct consequence of metastatic progression [1]. Other than aberrant cell proliferation, there is not a single more unifying phenotype in cancer biology.
Cancer occurs after cells in a tissue progressively acquire aberrant mutations to produce progeny with uncontrolled capacities of proliferation [2,3]. This uncontrolled mitosis produces a primary tumor which eventually undergoes metaplasia, followed by dysplasia and then anaplasia, resulting in a malignant phenotype. It is this malignant phenotype that provides a mechanism for intravasation into the circulation, followed by extravasation to a secondary site for tumorigenesis. Such a phenomenon is referred to as metastasis, defined as the spread of a cancer variant from one organ to another non-adjacent organ [4]. As opposed to invasion which involves the migration of cancer cells to adjacent tissue, metastasis necessitates the development of cells capable of surviving highly variable environments as they fragment from the primary tumor and migrate into the vasculature.
Most cancers are of epithelial origin, referred to as carcinomas. Epithelial cells are normally held in place by junctions to adjacent cells as well as the basal lamina matrix [3]. Most epithelial cells require such attachments in order to survive, thereby requiring carcinomas to undergo a series of mutations that not only remove this need of cell signaling, but allow the cell to detach from the lamina foundation. However, not all cancers face such an uphill battle. Leukemia is a highly heterogenic cancer of dedifferentiated leukocytes that causes more disease-related deaths of children (younger than 18 years of age) than any other disease in the United States [5]. The statistic includes all diseases and is a sobering reminder of the prevalence of childhood cancer, despite the commendable advancements in cancer therapy. Unlike carcinomas, leukemia has inherent cell motility as leukocytes are required to move throughout the vascular system, suggesting that no mutations are required for anchorage independent growth. As such, it seems likely that leukemias are inherently metastatic, endowed with cancer’s deadliest phenotype simply due to cell of origin. However, its most devastating characteristic may also be exposed as a fatal flaw if chemotherapeutic approaches are able to exploit the ease at which dedifferentiated leukocytes traverse the cardiovascular and lymphatic systems. This article will present the biology of metastasis development and how leukemia cells are inherently endowed with this devastating phenotype. Finally, it will be elucidated how clinicians may be able to exploit the motility of leukemia and metastatic emboli of other cancer types through an approach known as Sonodynamic Therapy (SDT), a treatment modality that combines chemotherapeutic agents with ultrasonic irradiation to preferentially damage malignant cells.
Although it may seem archaic by contemporary standards, the existence of metastasis was accurately predicted by Stephen Paget in his article, “The Distribution of Secondary Growths In Cancer of The Breast,” published in a 1889 volume of The Lancet. In the article, Paget examined postmortem data that had been assembled from 735 women with breast cancer and noted that the organ distribution of cancer cells in these patients followed particular migratory patterns. Due to these observations, Paget suggested that the movement of malignancy is not due to chance events; rather some tumor cells (the “seed”) grew preferentially in the microenvironment of select organs (the “soil”) and that migration to secondary sites resulted only when the appropriate seed was implanted in its suitable soil [6]. Paget's assertion that the microenvironment plays a critical role in regulating the growth of metastases has since been supported by countless experimental studies; a remarkable insight for the time that has since dramatically improved the understanding of cancer biology.
In order for normal cells to transition into malignant potency, there are several crucial aberrations that need to occur. Such characteristics include mitosis in the absence of external growth stimulatory signals, substantial growth in spite of exogenous inhibitory signals, angiogenesis (excluding various hematological malignancies), the potentiation of cell immortalization, and finally, the capacity of invasion and metastasis [2]. This is typically the last step in malignancy and is the phenotypic characteristic that kills most patients [7]. It should be noted that there is a considerable difference between invasion and metastasis. Invasion refers to the ability to thrust aside and displace adjacent tissues, while metastasis is the fragmenting of tumor cells from a primary tumor that enables migration to secondary sites via the circulatory system [3,8]. In other words, primary tumors invade adjacent tissue systems, while secondary tumors invade distant tissue systems through the settlement of migratory emboli. Metastasis of carcinomas and sarcomas (cells of connective tissue origin) consists of 5 consecutive steps which will be described in explicit detail:invasion and migration, intravasation, circulation, extravasation, and finally, the combination of colonization, proliferation and angiogenesis at the secondary tumor site (Figure 1). These steps will later be compared to the inherent metastasis of leukemia to denote similarities and differences in migratory patterns. It should be noted that while metastatic spread through the cardiovascular or lymphatic systems are the most common migratory routes, cells can also move in a transcoelomic manner in which the spread of a malignancy into body cavities can occur via seeding the surface of the peritoneal, pleural, pericardial, or subarachnoid spaces. Examples of transcoelomic activity include ovarian tumors that spread transperitoneally to the surface of the liver as well as mesothelioma and primary lung cancers migrating through the pleural cavity, causing malignant pleural effusion [9]. Additionally, the accidental transfer of cancer cells during surgical intervention is an artificial form of metastasis that presents an ever posing threat to patients in the clinic (Figure 1).
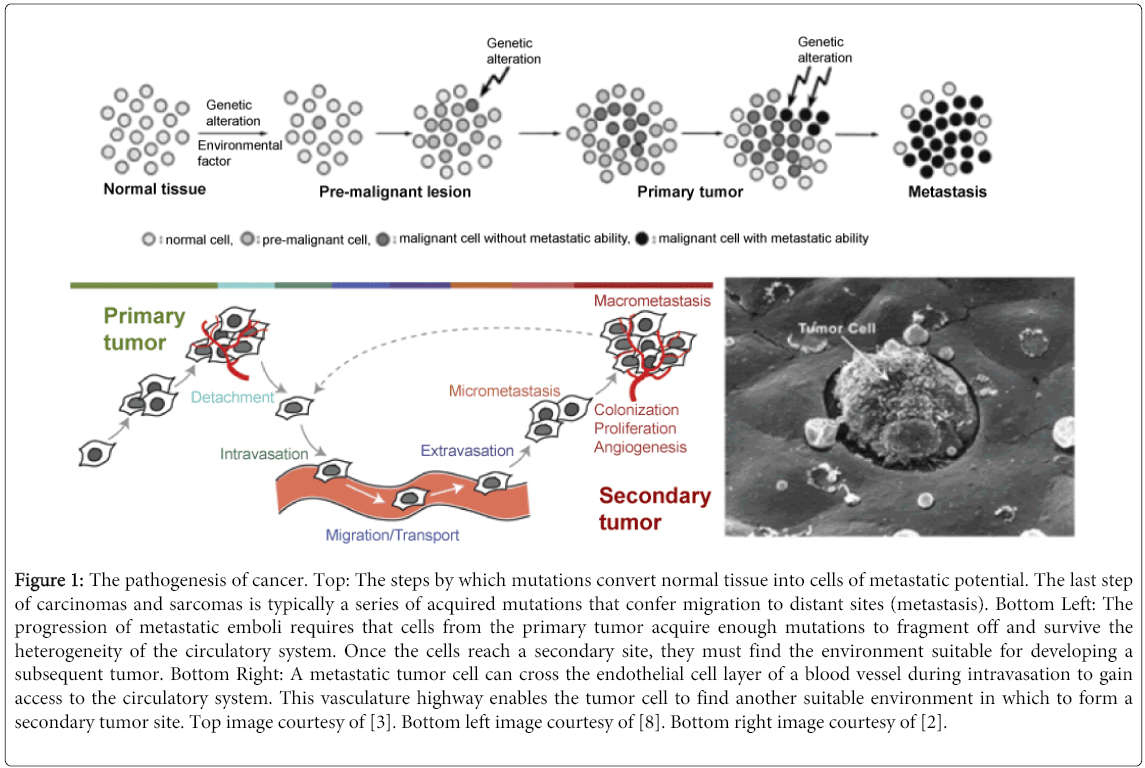
Figure 1: The pathogenesis of cancer. Top: The steps by which mutations convert normal tissue into cells of metastatic potential. The last step of carcinomas and sarcomas is typically a series of acquired mutations that confer migration to distant sites (metastasis). Bottom Left: The progression of metastatic emboli requires that cells from the primary tumor acquire enough mutations to fragment off and survive the heterogeneity of the circulatory system. Once the cells reach a secondary site, they must find the environment suitable for developing a subsequent tumor. Bottom Right: A metastatic tumor cell can cross the endothelial cell layer of a blood vessel during intravasation to gain access to the circulatory system. This vasculature highway enables the tumor cell to find another suitable environment in which to form a secondary tumor site. Top image courtesy of [3]. Bottom left image courtesy of [8]. Bottom right image courtesy of [2].
Invasion and Migration
In order for a primary tumor to become metastatic, neoplastic cell dissemination to different organs is an inherent necessity. Dissemination is a complex cell motility phenomenon, requiring the molecular coordination of protrusion, chemotaxis, invasion and contractility, all in an effort to achieve directed cell migration. The Epithelial-meshenchymal Mransition (EMT) is an important early step in carcinomas for the conversion of tumor cells into a migratory population that is capable of systemic metastasis [9,10]. In effect, epithelial cells lose their cell polarity and cell-cell adhesion as they transform into Mesenchymal stem cells (MSCs), gaining migratory and invasive properties of such cell types. When the cell reaches its target secondary site, it then reverts using the Mesenchymal-epithelial Transition (MET), allowing the carcinoma to seed in a distant organ. Although the EMT/MET transition is a natural phenomenon found in wound healing, it is aberrantly overused in tumorigenic epithelial cells [11-14].
Loss of E-cadherin is considered to be a fundamental event in EMT. Many Transcription Factors (TFs) that can repress E-cadherin directly or indirectly can be considered as some TFs bind to E-cadherin promoter and repress transcription. For example, SNAIL and ZEB factors bind to the E-box consensus sequences on the promoter region, while KLF8 binds to promoter through GT nucleotide boxes. These EMT-TFs not only directly repress E-cadherin, but also repress transcriptionally other junctional proteins found on epithelial cells, including claudins and desmosomes, facilitating EMT. Since EMT in cancer progression recaptures the EMT often found in development, many of the EMT-TFs are involved in promoting metastasis [14]. EMT also provides a substantial benefit to carcinomas in that it confers resistance to oncogene-induced premature senescence. TGFβ promotes tumor invasion as well as evasion of immune surveillance at advanced stages of malignancy. When TGFβ acts on activated Ras-expressing mammary epithelial cells, EMT is favored and apoptosis is inhibited [15]. EMT has even been linked to drug resistance as gain of EMT markers were found to be associated with the resistance of ovarian carcinoma epithelial cell lines to paclitaxel [16].
There is growing evidence that the formation of invadopodia is part of the EMT process [17,18]. Invadopodia are invasive actin polymerization-dependent protrusions found on cancer cells that degrade the Extracellular Matrix (ECM) separating cancer cells from the endothelium [10]. When plated on 2D surfaces, tumor cells have the propensity to form invadopodia on their ventral surfaces which degrade ECM through the delivery of metallo proteases. It has been shown that the formation of invadopodia is a multistep process involving actin-binding proteins and complex signaling cascades [10]. However, invadopodia also possess chemotaxis activity, allowing the tumor cells to move preferentially towards a favorable chemical gradient. There have been several studies that have observed human tumor cells migrating in 3D matrices in which the matrix-degrading invadopodia localized to the leading edge of the invading cell where the secretory machinery and metallo proteases were located [17-19].
In order for the metastatic cells to traverse distance between the epithelial lining and the endothelium leading up to intravasation, the development of substantial motility stands out as a necessity. High resolution multiphoton imaging of tumors in vivo has shown that malignant cells use both collective and single cell motility in their migration to the circulatory system. Breast carcinoma cell motility is characterized by solitary amoeboid movement at speeds significantly higher than other types of cell motility (~4 μm/min) [10]. The explanation to such high rates of travel is truly profound; invasive tumor cells form pseudopodia in vivo in response to Epidermal Growth Factor (EGF) secreted by tumor associated macrophages, as part of the tumor cell/macrophage paracrine loop described in breast tumors [20,21]. F-actin rich pseudopodia are the defining morphological feature of fast moving amoeboid cells and are involved in chemotaxis to direct tumor cells toward blood vessels before intravasation (Figure 2) [10].
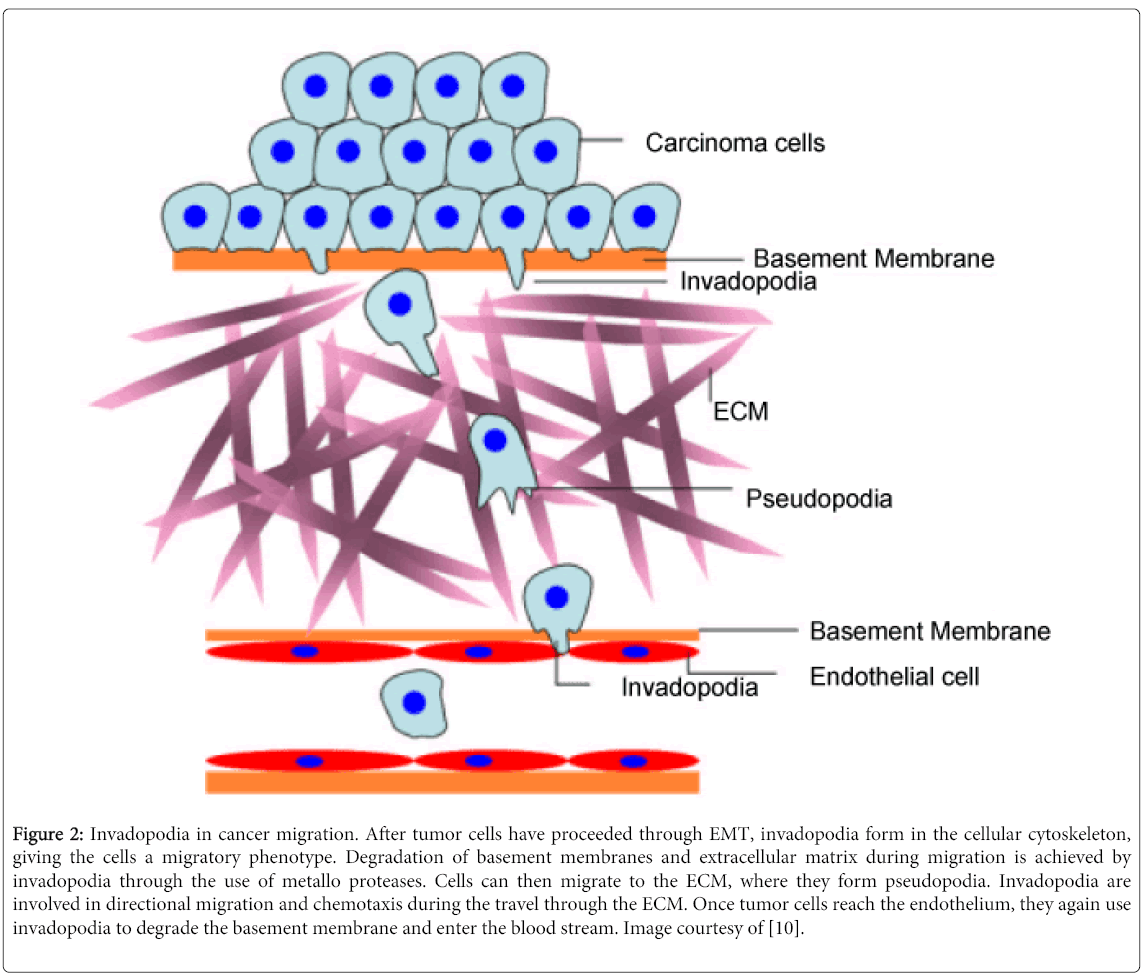
Figure 2: Invadopodia in cancer migration. After tumor cells have proceeded through EMT, invadopodia form in the cellular cytoskeleton, giving the cells a migratory phenotype. Degradation of basement membranes and extracellular matrix during migration is achieved by invadopodia through the use of metallo proteases. Cells can then migrate to the ECM, where they form pseudopodia. Invadopodia are involved in directional migration and chemotaxis during the travel through the ECM. Once tumor cells reach the endothelium, they again use invadopodia to degrade the basement membrane and enter the blood stream. Image courtesy of [10].
A major focus of current tumor biological research is the study of factors that regulate tumor cell migration. G-protein Coupled Receptors (GPCRs) bind chemokines and neuro transmitters and are the most prominent of these factors [11,12]. Although neurotransmitters are more associated with nerve signaling, they have been identified to have an impact on tumor cell migration. The migration of breast, prostate, and colon carcinoma cells are all enhanced by the stress-related neurotransmitter norepinephrine in vitro which is inhibited by the application of the β-blocker propranolol [11]. It has even been shown in vivo that the development of lumbar lymph node metastases in athymic BALB/c nude mice increased with the application of norepinephrine via micro-osmotic pumps, while propranolol inhibited this effect [22]. Such results suggest that stress levels may in fact play a pivotal role in tumor regulation, a concept that may have profound clinical applications.
Intravasation
Once metastatic carcinoma cells successfully detach from their epithelial lining, they must gain access to the circulatory system if they are to invade nonadjacent organ systems. While extracellular proteases degrade the ECM, establishing a basis for invasion, the tumor cells themselves must have the capacity to migrate down this path to reach the endothelial lining of the circulatory system. The macromolecular explanations for the acquisition of cell motility are rather straight forward as the remodeling of the actin cytoskeleton, along with the formation and dissolving of adhesion complexes are integral to generating this novel phenotype. As a result of the EMT-MET transition, the malignant cells are capable of developing lamellipodia that stretch in the moving direction [2,23]. On the front end of the cell, integrins build new focal contacts between ECM and lamellipodium, while the connections on the end are dissolved. By this approach, the cell moves and continuously secretes proteases on its march to the endothelium. On the edge of a lamellipodium, spiky structures known as filopodia, also form by the reorganization of actin fibers [23]. The formation of filopodia is integral for cell motility as they form focal adhesions with the substratum, linking it to the cell surface. Migrating cells almost always display filopodia as they are involved in the sensation of chemotaxis, resulting in changes to directed locomotion [24].
As the organization of cell shape as well as motility is subject to supervision by the protein family of Rho GTPases, the proteins are of monumental importance to intravasation. First and foremost, Rho proteins control contraction of the cell body. Such contractions pull the rear part of the cell in the moving direction, initiating movement [25]. Rho proteins can be categorized as Rho, Rac or Cdc42. Rho proteins are lipoproteins carrying a lipid attachment on their C-terminus, allowing for association with biomembranes [2,24,25].
The activation of special Receptor Tyrosine Kinases (RTKs) by growth factors ultimately results in the activation of many Rho proteins. This is readily apparent in the application of Platelet-derived Growth Factor (PDGF), a potent mitogen for cells of mesenchymal origin (including carcinomas in the mesenchymal state).Through the phosphorylation of PDGF receptor, several downstream effectors such as Raf, Ral-GEF and Phosphatidylinositol-3-kinase (PI3K) are stimulated via Ras activation. PI3K is of monumental importance for cell motility as it activates members of the Rho G-proteins via specific Guanine nucleotide Exchange Factors (GEFs), further perpetuating any aberrant signaling [25]. Rac and Cdc42 are crucial for intravasation as they induce the expression of many MMPs. Cdc42 is also capable of inducing filopodia and conveys stimulating effects on cell motility [2].
Chemokines are also involved in determining the direction of migration of various cells, including leukocytes and malignant cells. They bind to G-protein linked receptors and are divided into the following four subgroups: CXCL, CCL, CX3CL and CL [26]. In the context of cancer, chemokines represent important directional cues for invasion and metastasis. There are special chemokine receptors that enhance organ specificity of metastatic cells, thereby increasing the rate at which metastases successfully migrate to a compatible area. CXCR4 is present on the surface of malignant breast cancer cells and ligands of this receptor can be found on the surface of liver, lung and bone cells [27]. As expected, an increased amount of metastasis to these organs is observed. In addition, CXCR4 has shown to be vital for the lymph node specific metastasis of melanomas and ovarian cancers [28]. Due to their importance in metastatic spread, it is likely that antibody-based depletion of chemokines could be a viable therapeutic approach. In fact, it has been shown that IL-8 (angiogenic chemokine) monoclonal antibodies significantly inhibited tumor vascularization and growth in SCID mice with non-small cell lung cancer (NSCLC) [29].
Although cell motility is integral for intravasation, it is not until the malignant cells break through the lumen of lymphatic or blood vessels is this step of metastasis complete. Malignant cells can either directly or indirectly reach the cardiovascular system as many metastatic spreads initially begin in the lymphatic system. While classical cancer biology typically focuses on intravasation of hematological origin, it has been elucidated that lymphatic spread is just as relevant to metastasis. Integral to the purification of blood, the lymphatic system collects the interstitial fluid, moving the fluid through the lymph nodes and lymphatic vessels until it returns to cardiovascular circulation. It has even been proposed that carcinomas often spread initially via the lymphogenous system, whereas sarcomas tend to spread via the hematological system. This may be due to the fact that the penetration of lymphatic vessels is easier due to the absence of a continuous basement membrane and a coating layer of pericytes, as well as their weak inter-endothelial connections [30]. At the border of tumor and stroma, enlarged lymphatic vessels are typically observed, the formation being a consequence of the VEGF-C expression that drives lymphangiogenesis. Tumors with enhanced VEGF-C expression exhibit an increased metastatic potential, especially towards the draining lymph nodes [31]. In order to penetrate either the lymphatic or cardiovascular system, several proteases (such as MMP-1) are secreted which degrade the endothelium [32]. Migration to the vessels is facilitated with the help of several chemokines as well as altered expression of several adhesion molecules [2].
Circulation
Cells that manage to break through the endothelium of the circulatory system gain access to a vast network of vessels, substantially increasing the likelihood of secondary tumors. Under normal physiological conditions, most cells activate a special form of apoptosis known anoikis in the case they are unable to adhere to solid substrate or other cells (Figure 3) [33]. In fact, cancer cells that are still anchorage dependent at the time of entering the circulation have anoikis activated, thereby destroying the cells [34]. While most cells would not survive and proliferate in the heterogeneous environment of the vasculature due to the lack of a solid substrate, malignant cells typically acquire enough mutations for anchorage independent growth. In addition, if malignant cells are not yet independent of exogenous growth factors which are secreted by stroma cells, apoptosis is likely to be induced due to the absence of such growth factors in circulation [3].
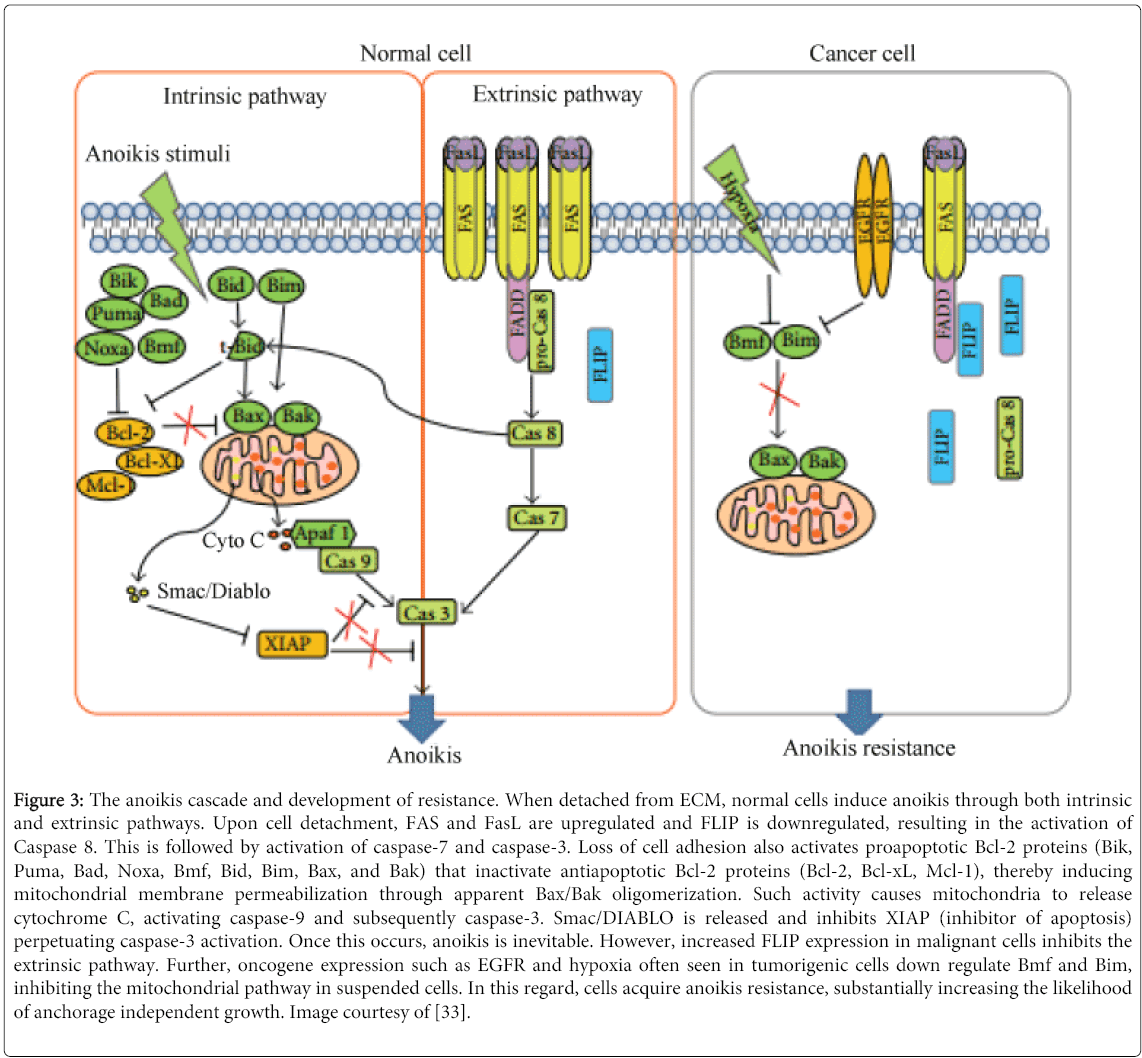
Figure 3: The anoikis cascade and development of resistance. When detached from ECM, normal cells induce anoikis through both intrinsic and extrinsic pathways. Upon cell detachment, FAS and FasL are upregulated and FLIP is downregulated, resulting in the activation of Caspase 8. This is followed by activation of caspase-7 and caspase-3. Loss of cell adhesion also activates proapoptotic Bcl-2 proteins (Bik, Puma, Bad, Noxa, Bmf, Bid, Bim, Bax, and Bak) that inactivate antiapoptotic Bcl-2 proteins (Bcl-2, Bcl-xL, Mcl-1), thereby inducing mitochondrial membrane permeabilization through apparent Bax/Bak oligomerization. Such activity causes mitochondria to release cytochrome C, activating caspase-9 and subsequently caspase-3. Smac/DIABLO is released and inhibits XIAP (inhibitor of apoptosis) perpetuating caspase-3 activation. Once this occurs, anoikis is inevitable. However, increased FLIP expression in malignant cells inhibits the extrinsic pathway. Further, oncogene expression such as EGFR and hypoxia often seen in tumorigenic cells down regulate Bmf and Bim, inhibiting the mitochondrial pathway in suspended cells. In this regard, cells acquire anoikis resistance, substantially increasing the likelihood of anchorage independent growth. Image courtesy of [33].
Even when malignant cells have acquired these advantageous adaptions, they are still highly vulnerable in the vasculature. In smaller vessels such as the arterioles or capillaries, high shear forces often occur. This can cause significant damage to neoplastic cells as they do not have the unique cytoskeletal structure of erythrocytes that permits such mechanical stress [3]. Such forces are likely to be a primary reason for metastatic inefficiency. Further, the heterogenic environment of circulation, characterized by extremely high concentrations of oxygen and lymphocytes, substantially impair the progression of malignant cells. Therefore, the presence of those adverse conditions actually provides a breeding ground for particularly aggressive and resistant cancer cells due to selection pressures [2]. Malignant cells antagonize the potentially toxic conditions of the vasculature by forming microemboli, an aggregation of tumor cells with thrombocytes and erythrocytes. The formation of microemboli is mainly due to tissue factor, a protein highly expressed in malignant carcinoma cells, but not on benign tumor or healthy epithelial cells. Tissue factor interacts with special proteins in the plasma to activate thrombin which converts fibrinogen into fibrin, thereby resulting in cell conglomeration [7]. The importance of metastatic emboli has been analyzed in knock-out mice which lacked specific components of the clumping-cascade. Melanoma cells were administered intravenously into the knock-out mice to assess metastatic efficiency. In comparison to the control group, metastasis was decreased by >90% [35]. By shielding neoplastic cells from the unstable environment of circulation, emboli formation becomes an integral mechanism for the survival of cancer metastases as they continue their migration towards a secondary location.
Extravasation
Eventually, partially due to chemokine signaling, partially due to trial and error, the metastatic cells approach an organ that is suitable for secondary growth. Before reaching that organ, the cells must find a way to exit the circulatory highway. If cancer cells survive the adverse conditions in the vessels and reach larger venous blood vessels, they are carried further by the bloodstream until they enter the capillary network of the lungs. There is a high probability that the cancer cells will become entangled in the capillary system of the lungs due to the sheer difference in size between malignant cells and the small vessels that must be successfully traversed. For comparison, cancer cells are often ~20 μm in size, in relation to the capillaries that are ~3-8 μm [2]. Unlike erythrocytes that are designed to meander through the narrow passages, malignant cells are not very elastic and often form microthrombi. Therefore, the probability that the metastatic emboli will become lodged in the arterioles is very high. It is nothing short of miraculous that any malignant cells are able to successfully traverse the circulatory system. Unfortunately for the patient, even a highly unlikely event becomes probable when enough cells reach this metastatic state.
Of course, the ever resilient cancer cells have mechanisms to greatly increase the odds in their favor. It is likely that neoplastic cells nearing the lung capillaries discard a tremendous amount of cytoplasm in order to form smaller, but still viable cells that are capable of passing through the small vascular network. It has also been elucidated that some cancer cells can avoid the capillary network by using arterial-venous shunts, bypassing the capillaries altogether [7]. Either way, when the malignant cells leave the lung capillaries and reach the general arterial vessels, they have access to a substantial variety of body tissue.
Once the cells have reached a favorable secondary site, extravasation will occur in two different ways. One possibility is that neoplastic cells begin proliferating in the lumen of a vessel. The rapidly growing tumor will eventually compromise the vessel wall, thereby providing an accessible route into the tissue [7]. The second possibility for malignant cells to penetrate an organ is similar to intravasation and includes the degradation of endothelium and basement membrane through proteolyic enzymes. Leukocytes also have the need to extravasate in great quantity over a short period in order to combat infection [36].
In the course of this process known as diapedesis, leukocytes adhere via selectins to the endothelium and perform a form of rolling adhesion to interact with endothelial cells, which in turn enables the passage of leukocytes. This is due to the fact that carbohydrate ligands on the circulating leukocytes bind to selectin molecules on the inner wall of the vessel, with marginal affinity. This enables leukocytes to slow down and begin rolling along the inner surface of the vessel wall (Figure 4) [36]. During this rolling motion, transitory bonds are formed and broken between selectins and their associated ligands, causing leukocyte motion to halt. Diapedesis involves a variety of molecules including LFA-1 + ICAM-2, CD3 + ICAM-1, as well as PECAM (CD31) and the complete process takes place in less than a minute [37].
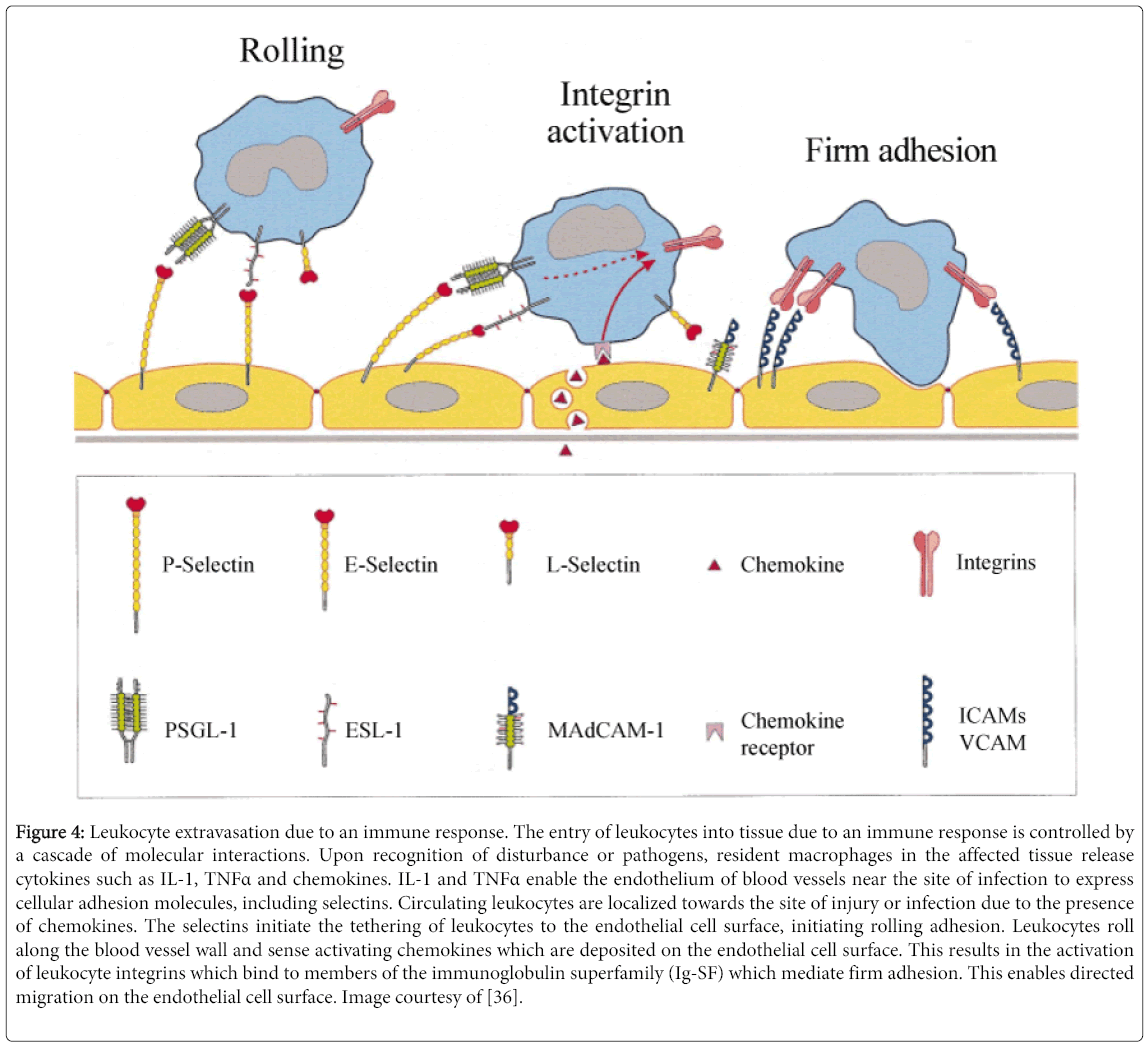
Figure 4: Leukocyte extravasation due to an immune response. The entry of leukocytes into tissue due to an immune response is controlled by a cascade of molecular interactions. Upon recognition of disturbance or pathogens, resident macrophages in the affected tissue release cytokines such as IL-1, TNFα and chemokines. IL-1 and TNFα enable the endothelium of blood vessels near the site of infection to express cellular adhesion molecules, including selectins. Circulating leukocytes are localized towards the site of injury or infection due to the presence of chemokines. The selectins initiate the tethering of leukocytes to the endothelial cell surface, initiating rolling adhesion. Leukocytes roll along the blood vessel wall and sense activating chemokines which are deposited on the endothelial cell surface. This results in the activation of leukocyte integrins which bind to members of the immunoglobulin superfamily (Ig-SF) which mediate firm adhesion. This enables directed migration on the endothelial cell surface. Image courtesy of [36].
Colonization, Proliferation and Angiogenesis at the Secondary Tumor Site
Now that the metastatic cells have successfully reached an ideal secondary location, they must quickly begin altering the extracellular environment within the organ to establish favorable conditions for a proliferating tumor. The new microenvironment typically does not provide the same survival and growth factors as the original tissue, which enabled the primary tumor to proliferate [7]. Without the special physiological factors of the original site, many metastasizing cells die or reside without proliferation as micrometastases. In fact, the amount of senescent micrometastases in cancer patients usually exceeds the amount of micrometastases that grow to a detectable size [2]. The detection of cytokeratin positive found exclusively on epithelial cells in completely mesenchymal tissues provides evidence for micrometastases. This technique enables physicians to track down single cell micrometastases amongst 105-106 surrounding mesenchymal cells [7]. It has been confirmed that the EMT which takes place at the beginning of metastatic invasion is reversed during colonization (referred to as MET) [38]. This transition enables metastasized tumor cells to adopt their original epithelial character.
A considerable amount of evidence indicates that molecular factors present in specific organs can influence whether or not metastatic cells will find the environment favorable for colonization [39]. Studies indicate that tumor cells will respond quite differently (gene expression, growth ability, and responsiveness to therapy) depending on the microenvironment of the new organ. Breast and prostate cells often metastasize to bone due to favorable interactions between the malignant and bone cells (osteoblasts and osteoclasts in particular). Molecules such as parathyroid hormone related protein and TGF-β are produced by the bone microenvironment, helping to satisfy the increased growth factor needs of a developing tumor [40]. The prototypical seed and soil situation that Paget proposed many years ago is also found in colon carcinomas and other cancers that metastasize to the liver. Specific growth factor receptors such as EGFR are found frequently on such cells as the corresponding growth factor is readily available in the hepatic microenvironment. Other growth factors such as TGF-a are commonly found in the liver and help regulate phenotypic characteristics of the tumor. This has been reflected by studies in which colorectal cancer cells of varying metastatic potential were planted directly into the liver, as growth regulation in the hepatic microenvironment actually determined several important tumorigenic characteristics of the malignant cells [41].
After initial colonization and proliferation of secondary tumors, there needs to be development of an adequate blood supply that can sustain the ever increasing metabolic activity of the tumorigenic growth. Since there are usually not enough adjacent blood vessels to recruit, tumors simply make their own. Angiogenesis is the formation of new blood vessels in the direction of a tumor to provide it with nutrients and to remove metabolic waste products [42,43]. Commonly seen in primary tumors, angiogenesis becomes just as integral to secondary malignancies that exceed the natural blood supply of their microenvironment. The initial growth of a tumor prior to angiogenesis is referred to as the avascular phase. Not until entering the vascular phase is a tumor capable of exponential proliferation. An angiogenic switch regulates the transition from avascular to vascular through the balance of pro and antiangiogenic factors. Angiogenic oncoproteins such as Ras can operate as angiogenic factors and enhance the expression of proangiogenic proteins, including VEGF, FGF (fibroblast growth factors), PDGF, and EGF [7].
The vascularization of a tumor is in stark contrast to the structure of normal vessels. In sum, it is more chaotic; often being enlarged, containing fragmented ends, uneven distribution of blood flow, and even holes that can eventually initiate hemorrhaging [44]. Existing angiogenesis inhibitors suppress either proliferation of the endothelial cells or attempt to induce apoptosis. Studies have also investigated angiogenic gene therapy in which adenoviral vectors are used for the transgenic expression of endostatin or other angiogenic inhibitors. Subsequent in vivo results indicate that tumor growth and metastasis could be mitigated, providing a potential avenue to treat metastatic progression [44].
Although cancers found more commonly in adults have particularly high mortality rates (pancreatic, liver, lung), they take much longer to develop malignancy than childhood cancers. In fact, many adult carcinomas require years of incubation before enough aberrant mutations have been acquired for detectable symptoms to become readily apparent [5]. By contrast, leukemia, the most prolific childhood cancer in the United States, often only takes a few months to spread throughout the patient. While it is true that leukemias are often found in adults, many of these cases are in the form of secondary leukemia, an unfortunate side effect of prior chemotherapeutic intervention [45,46]. In fact, leukemia has the highest proportion of child vs. adult malignancy rates than any other cancer [5]. It also should be noted that many of those adults fall within the 18-30 year old range, still substantially younger than most cancer incidence.
With such a high proportion of young individuals being diagnosed with leukemia, there must be some phenotypic characteristics that set the hematological malignancy apart from other cancers. This becomes readily apparent when the nature of leukocytes, the immune cells that dedifferentiate into leukemic malignancy, is taken into consideration. By default, leukocytes have profound mobility, circulating the vasculature in patrol or in response of pathogenic disturbance. Therefore, the first 2 steps of metastasis (invasion and migration, intravasation) are unnecessary as leukocytes are naturally found in systemic circulation. The classes of leukocytes are incredibly diverse by nature, as a result of being either of myeloid (monocytes and macrophages, neutrophils, basophils, eosinophils) or lymphoid (T-cells, B-cells and Natural Killer (NK) cells). Consequently, leukocytes give rise to a profound heterogeneity of cancers [47,48]. As expected, the demands of such a diverse cell type are satisfied by an ever-present population of Hematopoietic stem cells (HSCs). Cancers can be thought of cells that have been reprogrammed to a dedifferentiated state, marked by gene expression typically reserved for embryonic or adult stem cells. It is even necessary for leukocytes to maintain an innate form of extravasation as they need such characteristics to illicit a rapid immune response [36,37]. Taking all of these phenotypic characteristics together, it should come as no surprise why leukemias are the most prevalent childhood cancers in the United States. Based on the cancer biology of other cell types, it can be inferred that leukemia is endowed with inherent metastatic potential. In a sense, leukemias often require fewer mutations than cancers of other cell types to spread throughout the patient. It is this inherent metastasis that separates leukemia from any other malignancy.
Migratory Patterns of Leukemia
If leukemia was inherently metastatic, it should have characteristic migratory patterns as found in other cancers. Such an answer requires only a scalpel and patience (as well as patients).In fact, autopsy reports describing leukemia mortality have long been established in the clinical setting [49,50]. In most cases, it appears that leukemia has a similar seeding frequency of the skin, breast, trachea, diaphragm and all other muscles. Due to their cell type of origin, the highest incidence of metastasis is found in the lymphatic system (lymph nodes and spleen). The excess of metastases at specific sites do not cluster in topographical areas or in anatomical systems, with the exception of metastases in the central nervous and endocrine systems (acute lymphoblastic leukemia (ALL)). From the unique migratory patterns of ALL, it can be inferred that soil specificity may account for the higher than expected occurrence of metastases in the noted organs for a specific leukemia. Chronic Lymphocytic Leukemia (CLL) shows an excess of metastases in all lymph nodes, kidney, adrenals and heart. The unique seeding of CLL suggests a lymphatic route of dissemination, as opposed to a cardiovascular spread of malignant cells [49].
While more common leukemias such as ALL and CLL have been categorized for years, the unique migratory patterns of NK-derived leukemias were categorized much later [51,52]. Pathologically, NK leukemia shows variable cytological appearances, with frequent angiocentricity (angioinvasion and angiodestruction) associated with zonal necrosis of affected areas. Most cases occur in the nasopharynx and upper aerodigestive tract. However, occurrence in non-nasal sites such as the skin, gastrointestinal tract and testis is also observed [52]. These malignancies are also marked by tumor cells with NK cell characteristics, including the immunophenotype of CD2+, surface CD3-, cytoplasmic CD3ε+, CD7+/-, and CD56+. Identification of NK-derived malignant cells is furthered by the presence of the germline T cell receptor gene, inferring the cell type of origin. While NK leukemias have apparent differences in their migratory patterns, they share a common trait with other hematological malignancies of leukocyte origin in the formation of aberrant skin lesions (cutis) [52,53]. Leukemia cutis refers to the infiltration of neoplastic leukocytes or their precursors into the skin resulting in readily identifiable cutaneous lesions. Patient examination typically reveals multiple hyper-pigmented nodules and plaques involving the face, trunk, and extremities. A diagnosis of leukemia cutis generally portends a poor prognosis as it strongly correlates with additional sites of extramedullary involvement [53]. Unlike most cancers, leukemia has the capability to blanket the dermal layers with cutaneous lesions, providing a profound visual example of its metastatic extent.
Mixed Lineage Leukemia
Due to the extensive diversity of leukocytes, metastatic progression of several variants can result in Mixed Lineage Leukemia (MLL), a characteristic that presents a nightmare scenario in the clinic. MLL refers to a very aggressive hematological malignancy that predominates in pediatric patients [54]. In contrast to other types of childhood acute leukemias, MLL offers a dismal prognosis and despite the availability of advanced treatment methods, improvements in patient outcome have been modest at best. MLL is characterized by the presence of MLL fusion proteins that are the result of chromosomal translocations affecting the MLL gene at 11q23. The genetic aberrations are significant as the MLL protein (Histone-lysine N-methyltransferase) is a histone methyltransferase known to be a positive global regulator of gene transcription and is involved in the epigenetic maintenance of transcriptional memory. The resulting chimeras are transcriptional regulators that take control of targets normally controlled by MLL as evidenced by deregulation of clustered HOX homeobox genes (Figure 5) [55]. MLL has also been characterized by partial tandem duplications as well as amplifications, all of which perpetuate sustained HOX expression and stalled differentiation. MLL is appropriately named as lesions are associated with both Acute Myeloid Leukemia (AML) and ALL.
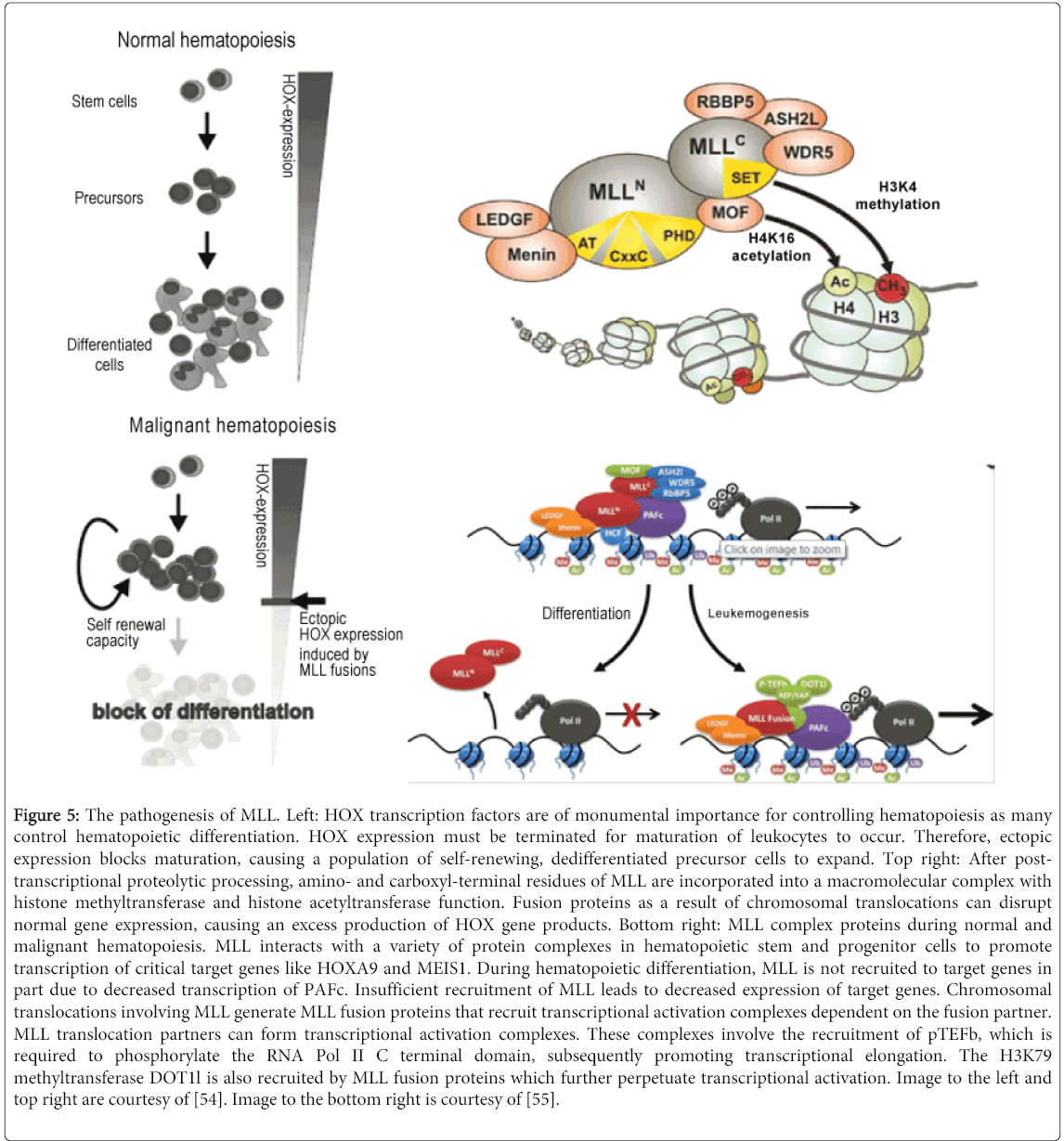
Figure 5: The pathogenesis of MLL. Left: HOX transcription factors are of monumental importance for controlling hematopoiesis as many control hematopoietic differentiation. HOX expression must be terminated for maturation of leukocytes to occur. Therefore, ectopic expression blocks maturation, causing a population of self-renewing, dedifferentiated precursor cells to expand. Top right: After posttranscriptional proteolytic processing, amino- and carboxyl-terminal residues of MLL are incorporated into a macromolecular complex with histone methyltransferase and histone acetyltransferase function. Fusion proteins as a result of chromosomal translocations can disrupt normal gene expression, causing an excess production of HOX gene products. Bottom right: MLL complex proteins during normal and malignant hematopoiesis. MLL interacts with a variety of protein complexes in hematopoietic stem and progenitor cells to promote transcription of critical target genes like HOXA9 and MEIS1. During hematopoietic differentiation, MLL is not recruited to target genes in part due to decreased transcription of PAFc. Insufficient recruitment of MLL leads to decreased expression of target genes. Chromosomal translocations involving MLL generate MLL fusion proteins that recruit transcriptional activation complexes dependent on the fusion partner. MLL translocation partners can form transcriptional activation complexes. These complexes involve the recruitment of pTEFb, which is required to phosphorylate the RNA Pol II C terminal domain, subsequently promoting transcriptional elongation. The H3K79 methyltransferase DOT1l is also recruited by MLL fusion proteins which further perpetuate transcriptional activation. Image to the left and top right are courtesy of [54]. Image to the bottom right is courtesy of [55].
The profound heterogeneity of MLL has been characterized in an attempt to further understand the disconcerting prognosis of patients diagnosed with the debilitating condition [56]. Despite immunological evidence of lymphoid differentiation (17 CALLA+ and one T cell-associated antigen+) and findings of immunoglobulin gene rearrangement, blasts from all patients reacted with one to five monoclonal antibodies identifying myeloid-associated cell surface antigens (My-1, MCS.2, Mo1, SJ-D1, or 5F1). Dual staining with microsphere-conjugated antibodies and analysis by flow cytometry further confirmed that some blasts were simultaneously expressing lymphoid and myeloid associated antigens, thereby validating the name given to the malignancy. With metastatic potential acquired from its leukocyte origin, MLL is free to rapidly proliferate throughout the patient, aggressively invading surrounding vasculature until the patient’s blood becomes overcrowded with dedifferentiated metastases, substantially depleting the availability of healthy blood cells [57].
Uncontrolled Hematopoietic Stem Cells Perpetuate Leukemia
Perhaps one of the most fundamental hallmarks of cancer is the transition into a dedifferentiated state that reflects phenotypic characteristics of stem cells. However, in the case of malignant cells, they no longer have the capacity to differentiate into a required cell type. Rather, they retain the phenotypic characteristics necessary for rapid proliferation and become trapped in an unending cell cycle. Of course, it is much easier for a progenitor stem cell lineage to become cancerous than fully differentiated cells, as considerably less mutations are required for tumorigenic growth and eventually metastatic potential. As is seen in leukemia, Hematopoietic Stem Cells (HSCs) play a monumental role in malignancy development. During hematopoiesis, the progenitor HSCs receive a complex array of transcription factors and growth signals to differentiate into all known existing blood cells. Within this system, HSCs give rise to either the lymphoid (adaptive immunity) or myeloid (innate immunity, megakaryocytes and erythoids) lineage as subsequent cell progeny are directed by further differentiation steps to give rise to the desired cell type. However, when improper signaling or mutations occur within HSCs, the likelihood of leukemia pathogenesis is dramatically increased [58]. Normal hematopoiesis requires complex interactions between the bone marrow microenvironment (the stem cell niche) and HSCs. These interactions are critical for the maintenance of normal HSC activity and deregulation of this intricate system aberrantly influences HSC self-renewal. If this self-renewal is left unchecked, progression into Leukemia Stem Cells (LSCs), that also possess limitless self-renewal, may be generated in the stem cell population, hijacking the homeostatic mechanisms necessary for proper differentiation. LSCs are extremely resilient as they take refuge within the sanctuary of the niche during chemotherapy, and consequently contribute to eventual disease relapse [48].
Such deregulation of HSCs is apparent in AML, a leukemia that has significant clinical relevance due to its prevalence in patients. AML is the second most common leukemia diagnosed in both adults and children, with a 5 year survival rate of only 24% [5]. As such, HSC deregulation leading to AML is a common phenomenon in the clinic and has been meticulously studied in order to further elucidate the disease’s pathogenesis. As for many hematological malignancies, AML has been extensively characterized as a cell autonomous disorder as the genetic events leading to transformation of the normal hematopoietic cell are found within that cell and are both necessary as well as sufficient for leukemic development. Aberrant fusion proteins, such as MLL-AF9 or MLL-ENL1 that are expressed as a consequence of chromosomal translocations are typically present in leukemic blasts derived from patients with AML [48]. Introduction of the subsequent alleles into normal bone marrow cells perpetuates AML in murine models of disease, validating their oncogenic properties. This constructed AML accurately recapitulates the human disease phenotype, including hallmark stem cell properties such as the ability to consecutively replate on methylcellulose in the absence of stroma and the ability to confer an AML phenotype that can be transplanted in vivo [59].
Although all AML cells are thought to harbor the cell-autonomous mutations that are found in its pathogenesis, the subpopulation of AML cells referred to as LSCs are responsible for the long-term repopulating potential as well as the ability to propagate and maintain the AML phenotype. It has been estimated that as few as one in a million AML cells possess leukemia initiating activity. Therefore, AML has a hierarchical organization similar to that of normal hematopoiesis in which there is a rare subpopulation of cells with limitless self-renewal potential that gives rise to progeny that lack such potential [60,61]. LSCs in certain types of murine AML (induced by MLL-AF9, 2 MOZ-TIF2, 2 MOZ-TIF3 or MLL-ENL1) have characteristics of progenitor cells with phenotypic characteristics similar to normal granulocyte/macrophage precursors. These leukemia-initiating cells are more mature than HSCs, but have acquired limitless self-renewal through oncogenic transformation, leading to the activation of essential stem cell genes [62,63]. Such observations are indeed profound as it suggests that only a relatively few amount of AML cells are responsible for perpetuating metastatic disease in the patient.
This concept opens up the idea of finding therapeutic approaches that specifically target leukemic progenitors, an approach that has been under considerable investigation (Figure 6) [62,64]. If treatments can be successfully devised, the malignant cells actually responsible for maintaining leukemia cell populations will be eradicated, effectively neutralizing disease progression.
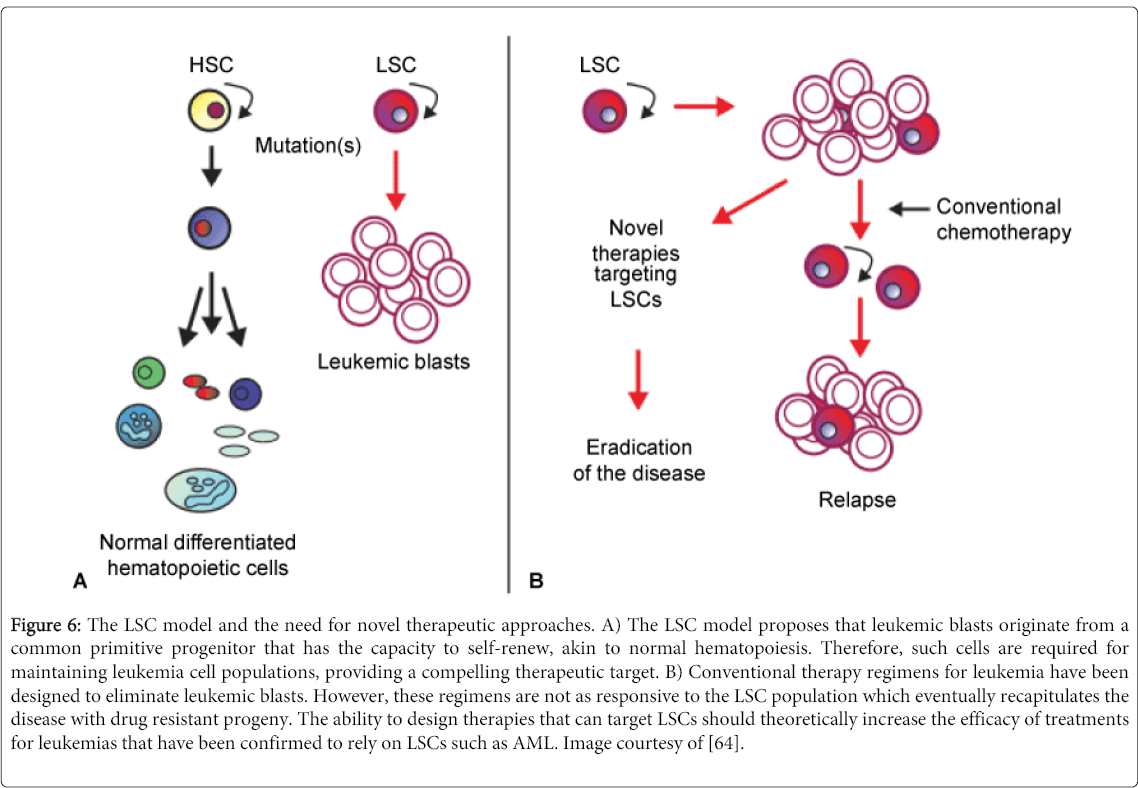
Figure 6: The LSC model and the need for novel therapeutic approaches. A) The LSC model proposes that leukemic blasts originate from a common primitive progenitor that has the capacity to self-renew, akin to normal hematopoiesis. Therefore, such cells are required for maintaining leukemia cell populations, providing a compelling therapeutic target. B) Conventional therapy regimens for leukemia have been designed to eliminate leukemic blasts. However, these regimens are not as responsive to the LSC population which eventually recapitulates the disease with drug resistant progeny. The ability to design therapies that can target LSCs should theoretically increase the efficacy of treatments for leukemias that have been confirmed to rely on LSCs such as AML. Image courtesy of [64].
Throughout this article, similarities between metastatic carcinomas and the inherent metastasis of leukemia have been proposed in order to suggest that leukemia cells require less aberrant mutations to spread throughout a patient.
This comparison has been made for a very practical reason; if metastatic emboli are comparable to leukemias, perhaps the unique phenotype that links the malignant growths can be exploited to improve current chemotherapeutic approaches. That is, use the malignancy’s most devastating phenotypic characteristic to develop novel therapeutic methods that specifically target and damage circulating cancer cells. Such an approach could not only significantly improve leukemia chemotherapy, but also treatments for carcinomas that commonly metastasize and form secondary tumors. After all, it is the metastatic phenotype of cancer that subdues most patients.
This form of targeted chemotherapy may be achieved by Sonodynamic Therapy (SDT), a novel treatment modality that uses ultrasonic irradiation with chemotherapeutic agents known as sonosensitizers to preferentially damage malignant cells. It has been shown in countless experiments that ultrasound preferentially damages malignant cells based on the size differential between such cells and those of normal histology [65-67]. This is especially important for leukemia and other metastatic diseases as the malignant cells will be in close proximity to normal blood cells. By targeting the malignant cell’s inherent size differential with normal cells in circulation, SDT asserts itself as a therapeutic approach that is both effective and specific. The size differential between malignant and normal cells can be dramatically increased through the use of sonosensitizers that specifically target malignant cells, thereby amplifying the preferential damage of ultrasound.
SDT has also been shown to be particularly effective in drug resistant cell lines. One of the most cited shortcomings of chemotherapy in clinical practices is drug resistance acquired by tumors. SDT has been shown to reverse this potent defense mechanism in human leukemia multidrug resistance cell line K562/A02 (Figure 7) [68]. Reversal of drug resistance has also been observed in HepG2/ADM resistant hepatocellular carcinoma cells, demonstrating the versatility of SDT [67]. Such drugs often attack cells through multiple mechanisms, creating a potential synergistic effect when sononosensitizers of different classes are used in collaborative efforts [65]. Being able to develop treatment regiments in which the synergistic effects of different sonosensitizers are applied can have monumental importance in clinical applications. Such treatments could substantially amplify the capability of ultrasound to preferentially damage malignant cells in order to decrease the rate at which drug resistance is observed.
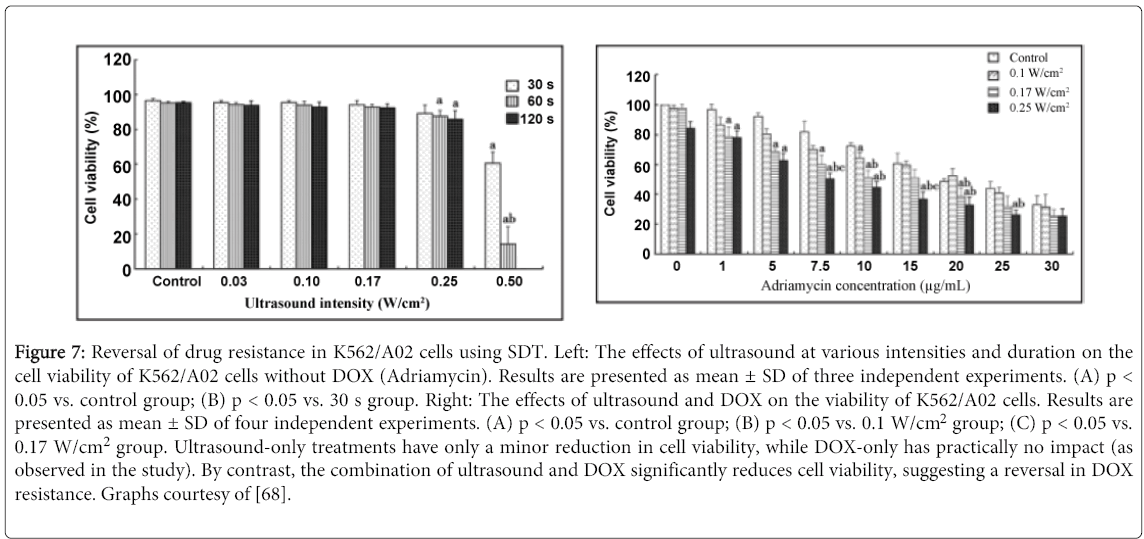
Figure 7: Reversal of drug resistance in K562/A02 cells using SDT. Left: The effects of ultrasound at various intensities and duration on the cell viability of K562/A02 cells without DOX (Adriamycin). Results are presented as mean ± SD of three independent experiments. (A) p < 0.05 vs. control group; (B) p < 0.05 vs. 30 s group. Right: The effects of ultrasound and DOX on the viability of K562/A02 cells. Results are presented as mean ± SD of four independent experiments. (A) p < 0.05 vs. control group; (B) p < 0.05 vs. 0.1 W/cm2 group; (C) p < 0.05 vs. 0.17 W/cm2 group. Ultrasound-only treatments have only a minor reduction in cell viability, while DOX-only has practically no impact (as observed in the study). By contrast, the combination of ultrasound and DOX significantly reduces cell viability, suggesting a reversal in DOX resistance. Graphs courtesy of [68].
In order to use SDT in the clinical setting, effective measures need to be devised in which to administer ultrasound as well as the chemotherapeutic agents to patients. Seeing that SDT has yet to be tested in the clinical setting, there has been no analysis as to how this treatment modality could be practically applied to patients. Although SDT fundamentally relies on an ultrasound system, there are a variety of ways in which the generated ultrasonic irradiation can be delivered. The most pragmatic approach that takes full advantage of leukemia’s inherent metastasis is a form of Extracorporeal Blood Sonication (EBS) in which blood is drawn out of the body, as in hemodialysis, and then sonicated before it reenters the patient. This approach will be described in explicit detail, along with cytochalasin B, a sonosensitizer that could have immediate clinical importance. It is hoped that such efforts will convince academics and clinicians of how the profound mobility of metastatic disease can be exploited to improve chemotherapeutic approaches in many clinical contexts.
Extracorporeal Blood Sonication
From everyday life, it is common knowledge that it is easier to hear music outside than through multiple walls. As such, ultrasonic irradiation loses some of its intensity as it travels through the human body. If there was a way to remove malignant cells from the body so they could be treated in an extracorporeal environment, there would be no sound inhibitors protecting such cells from the preferential damage of SDT. Although such an approach is unfeasible for most malignancies, leukemia is unique in that it does not form a primary tumor site. Rather, it flows through the blood, alongside normal cells as it slowly overcomes the natural defenses of the immune system. However, its most beneficial asset can be exploited to become a profound fatal flaw. Since most leukemias are localized in the blood, it would be rather straightforward to draw the malignant cells out their hiding place through dialysis. While dialysis is typically used on patients to act as an artificial replacement for lost kidney function due to renal failure, it could just as easily be used to treat leukemia in an extracorporeal setting. Sonosensitizers would be injected intravenously with some time passing before injection and sonication. The patient would then undergo a typical hemodialysis procedure in which blood is pumped outside of the body, thereby removing the natural sound barriers of human anatomy. There would be nothing standing in the way between the malignant cells and the ultrasonic waves that are able to inflict such profound preferential damage. In effect, this SDT procedure allows an in vivo setting to become almost in vitro. Since the in vitro studies of SDT with leukemia have yielded impressive results, this may be the most effective way to administer ultrasonic irradiation to such patients. However, it goes without saying that sound intensities used in EBS will have to be carefully monitored. There is very little standing in the way between the blood and the high intensity ultrasound being administered. While normal erythrocytes and leukocytes are more resistant to SDT, they are not invulnerable. Sufficient sound intensities will cause just as much damage to these cells as the malignant cells that are within close proximity [69]. Therefore, the sound intensity used in EBS would likely have to be considerably reduced. Nevertheless, it still provides the most direct route for sonicating the dedifferentiated blasts that cause so much devastation in patients. Further, there have been ultrasonic probes already devised that could be used to sonicate bone marrow containing aleukemic cells as well the LSCs shown to be the driving force behind many hematological malignancies. The combinatorial approach of EBS with such probes could provide a method to comprehensively sonicate almost all areas where metastatic disease (including carcinomas) may reside.
Cytochalasin B as the Prototypical Sonosensitizer
By default, malignant cells have a perturbed cytoskeleton due to the effects of dysplasia and subsequent anaplasia. With so many alterations present in malignant cells, the cytoskeleton provides an ideal opportunity for preferential damage. Specifically, one of the most intriguing possibilities is to preferentially lyse malignant cells based on their considerable size differential to cells of normal histology. This known fact invariably gives rise to the idea of grossly enlarging tumor cells to increase their already noticeable size difference with normal cells. Cytochalasin B is a known pharmacological agent that disrupts the actin cytoskeleton and inhibits cytokinesis by interfering with formation of the contractile ring as well as the development of the cleavage furrow [65]. Consequently, the cell does not divide and an immature actin cytoskeleton remains. However, the cell continues to form nuclei and eventually becomes grossly enlarged and multinucleated. Such cells invariably have more DNA targets, increasing the likelihood of apoptosis. Furthermore, the multinucleated cells have a large cell volume, making them more susceptible for direct cell destruction. Preferential damage of malignant cells is actually easily attainable as normal cells exposed to cytochalasin B exit the cell cycle and enter a resting state until sufficient actin levels are restored. Therefore, only malignant cells that have lost the ability to enter the rest phase will become grossly enlarged and multinucleated, providing an ideal target for ultrasonic irradiation. To put the size differentials into perspective, normal erythrocytes are typically between 6-8 µm and leukocytes range between 10-15 µm (the occasional macrophage grows up to 20 µm). By contrast, cytochalasin B treated U937 promyelocytic leukemia cells have been shown to grow in excess of 20 µm with some reaching nearly 40µm in diameter after enough exposure [70]. Such cells have reduced cytoskeletal integrity and are easy targets for ultrasonic irradiation.
Cytochalasin B also appears to have a profound impact on mitochondrial activity. When U937 promyelocytic leukemia cells were exposed to a relatively small concentration of Cytochalasin B (1.5 µM), the cells exhibited a 4-fold increase in comparison to U937 cells of typical histology, as assessed by MTT assay (Figure 8) [70]. Such a dramatic increase in metabolic rate inherently suggests using mitochondrial agents in tandem with cytochalasin B during ultrasound treatments. Indeed, Reactive Oxygen Species (ROS) agents often target the mitochondrial induced apoptotic pathway of leukemia cells, providing a viable method to develop synergistic treatments. This approach could be further supported by nucleic acid agents as cytochalasin B treated U937 cells are considerably multinucleated. It is very likely that only a single nucleus will have to undergo apoptosis in order to destroy the malignant cell; having so many nuclei present greatly increases the chances of this event.
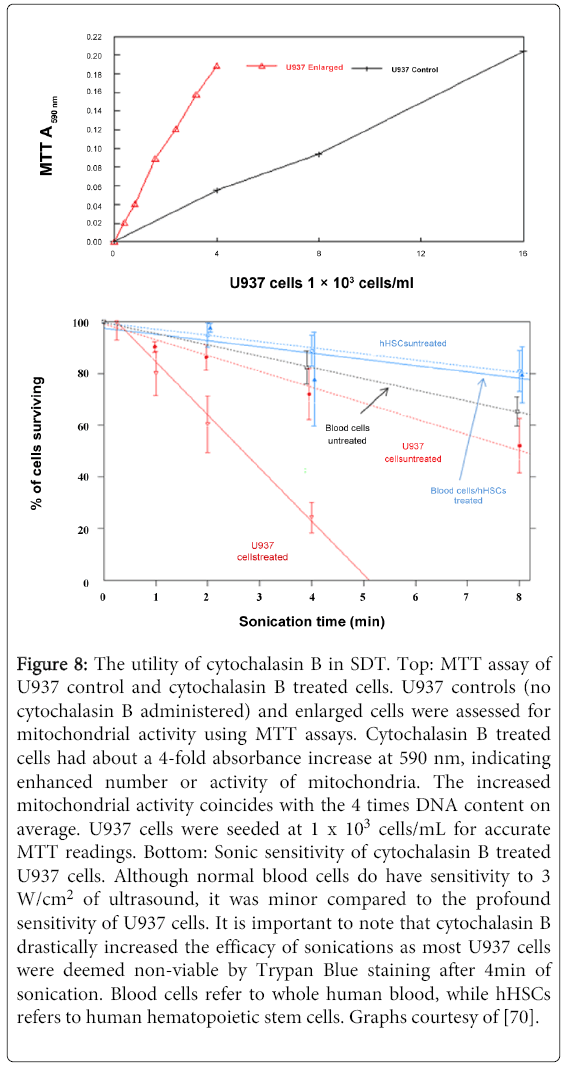
Figure 8: The utility of cytochalasin B in SDT. Top: MTT assay of U937 control and cytochalasin B treated cells. U937 controls (no cytochalasin B administered) and enlarged cells were assessed for mitochondrial activity using MTT assays. Cytochalasin B treated cells had about a 4-fold absorbance increase at 590 nm, indicating enhanced number or activity of mitochondria. The increased mitochondrial activity coincides with the 4 times DNA content on average. U937 cells were seeded at 1 x 103 cells/mL for accurate MTT readings. Bottom: Sonic sensitivity of cytochalasin B treated U937 cells. Although normal blood cells do have sensitivity to 3 W/cm2 of ultrasound, it was minor compared to the profound sensitivity of U937 cells. It is important to note that cytochalasin B drastically increased the efficacy of sonications as most U937 cells were deemed non-viable by Trypan Blue staining after 4min of sonication. Blood cells refer to whole human blood, while hHSCs refers to human hematopoietic stem cells. Graphs courtesy of [70].
HMME (Hematoporphyrin Monomethyl Ether)is a ROS agent that has been used for multiple cancer cell lines and has shown commendable efficacy, particularly in a study that involved U937 cells [71]. Immediately after administration, intracellular HMME concentrations rapidly increased within the U937 cells, reflecting its high affinity for malignant tissue. The synergistic effect of ultrasound with HMME showed significant cell destruction, vindicating the necessity of sonosensitizers in ultrasound mediated therapy (Figure 9). Flow cytometry with DCFH-DA (2'-7'-Dichlorodihydrofluorescein diacetate) staining confirmed that HMME-ultrasound treated cells had markedly increased ROS levels compared with the control, HMME and ultrasound-alone groups. Further analysis of damaged cell populations revealed that oxidative stress was present and that cells had indeed undergone apoptosis. These results not only confirm the linkage between ROS and apoptosis within U937 cells, but ultimately suggest a novel approach to treat leukemia patients. This damage can be further enhanced with the use of Doxorubicin (DOX) in collaboration with ultrasound/HMME as a study with QBC939 leukemia cells has indicated [72]. The study demonstrated that the ultrasound/DOX/HMME group had a higher reduction in cell viability than either the ultrasound/DOX or ultrasound/HMME groups, suggesting the need to investigate the cumulative effect of multiple sonosensitizers. Such results reflect a synergistic effect as DOX has the ability to increase ROS content in malignant cells. Since HMME and DOX have the capability to initiate increased production of singlet oxygen (ROS generated) when activated by ultrasonic irradiation, the drugs act in tandem to create a potent cytotoxic environment that malignant cells find particularly cytotoxic due to their decreased levels of endogenous thiol buffers.
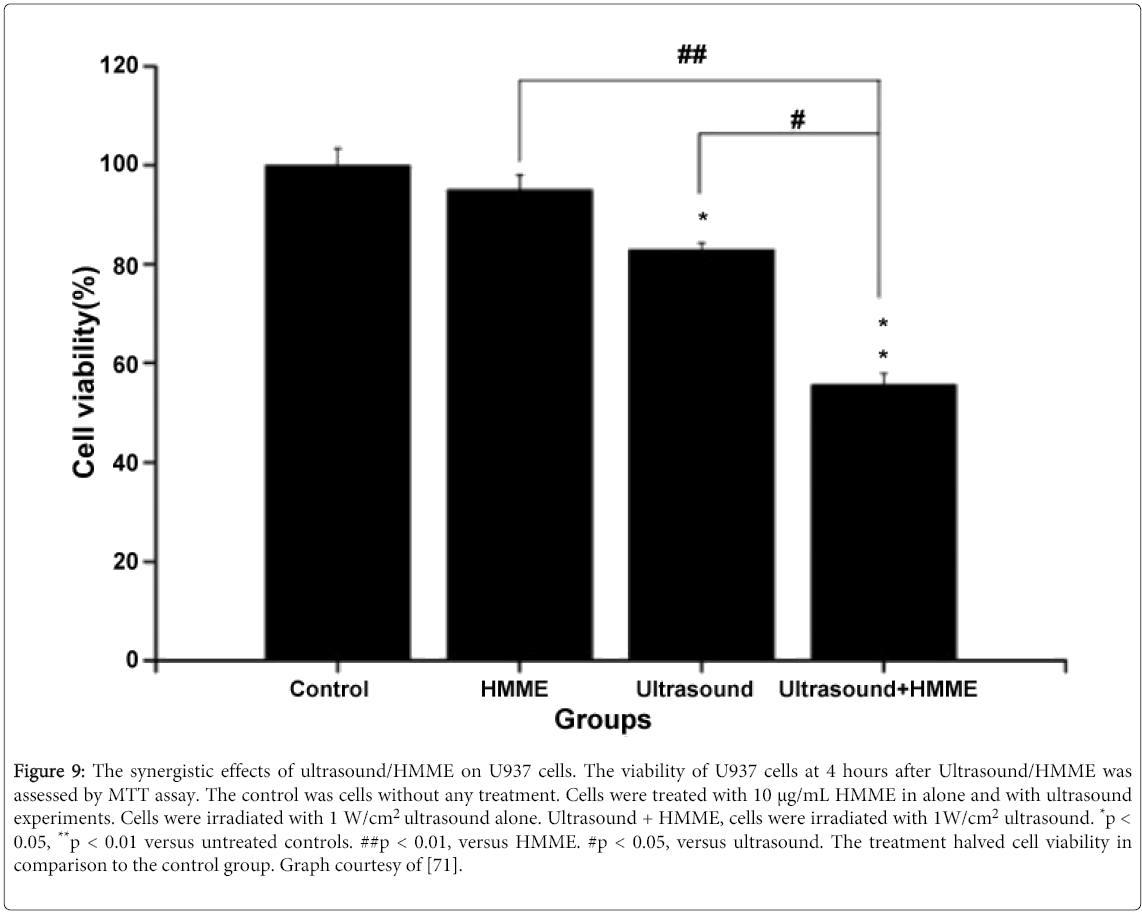
Figure 9: The synergistic effects of ultrasound/HMME on U937 cells. The viability of U937 cells at 4 hours after Ultrasound/HMME was assessed by MTT assay. The control was cells without any treatment. Cells were treated with 10 μg/mL HMME in alone and with ultrasound experiments. Cells were irradiated with 1 W/cm2 ultrasound alone. Ultrasound + HMME, cells were irradiated with 1W/cm2 ultrasound. *p < 0.05, **p < 0.01 versus untreated controls. ##p < 0.01, versus HMME. #p < 0.05, versus ultrasound. The treatment halved cell viability in comparison to the control group. Graph courtesy of [71].
The inherent metastasis of leukemia is readily apparent as it is derived from leukocytes that are designed to cover vast distances in the human body in a very short amount of time. While carcinomas and other cancers must acquire specific mutations to gain this profound mobility, leukemias need only become tumorigenic before being defined as a metastatic disease. Without having the need to develop mutations that permeate invasion and migration, followed by intravasation, these hematological malignancies are free to circulate throughout the body as malignant growths.
Further, leukocytes have an innate form of extravasation, suggesting it is less complicated for dedifferentiated blasts to escape the circulatory system. While leukemias do not form primary tumors, they are capable of rapidly crowding out healthy blood cells as their unchecked growth sequesters valuable resources. Eventually, the patient’s immune system will become entirely compromised as healthy leukocytes have been replaced by a sludge of white, dedifferentiated cells, the hallmark that gave leukemia its name (leukoshaima is white blood in Greek).
However, leukemia’s most beneficial phenotypic characteristic can be exploited by novel chemotherapeutic approaches. Specifically, SDT is capable of sonicating the circulatory highway that dedifferentiated leukocytes use to migrate towards distant tissue sites. Ultrasound can be used in tandem with chemotherapeutic agents that substantially amplify the preferential damage inflicted on malignant cells. As indicated by experimental evidence, ultrasonic waves produce remarkable antitumor effects under appropriate conditions. However, such effects are not always widespread and tumor populations often become resistant to ultrasound-only treatments. That is why SDT is such a sensible prospect as it significantly enhances the efficacy of ultrasonic irradiation, while still displaying preferential damage towards malignant cells. Every mechanism by which ultrasound destroys malignant tissue can in fact be amplified when an appropriate sonosensitizer is administered [65]. Such drugs often attack cells through multiple mechanisms as well, creating a potential synergistic effect when sonosensitizers of different classes are used in collaborative efforts. It has even been shown to reverse drug resistance in multiple cell lines, mitigating one of cancer’s most prolific defense mechanisms.
That is not to say all of the efforts put into developing SDT should be directed towards leukemia as there are other malignancies that take the lives of many more individuals each year. In fact, SDT could serve a tremendous utility in the clinic by eradicating metastatic emboli and micrometastases of other cancer types. Doing so might prevent metastatic growths from forming in the patient, allowing clinicians to focus on the primary tumor which can easily be treated with adjuvant therapy (pending on the performance status of the patient). It just should be noted that SDT has found particular promise with leukemia and that many lives could be saved if effective treatments are developed. At any rate, the idea of combining ultrasound with drugs that amplify the ways in which it preferentially damages malignant cells is gaining more legitimacy as successful studies have indicated the potential of SDT. By using the synergistic effects of ultrasonic irradiation and sonosensitizers, SDT is proving to be a viable treatment modality that has the capability to revolutionize the way in which chemotherapy is administered in the clinical setting. Such an approach exploits the inherent metastasis of leukemia, turning an implicitly advantageous phenotype into a fatal flaw. All that remains is the necessary clinical testing. Only through these necessary trials will enough evidence be compiled to conclude whether SDT is in fact as good as advertised.
The author would like to thank Syracuse University for their financial support of SDT research as it has served a tremendous utility in advancing the therapeutic approach.