Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 2
In this study, data mining approach was used to generate association rules for predicting average flexibility from the various derived sequence and structural features. 21 parameters were calculated and their variable importance was calculated for 115 sequences of AGC kinase family belonging to mouse and human using Classification and Regression Tree (CART). Beta turns were found to have maximum influence on average flexibility while the total beta strands were found to exert minimum impact on average flexibility. Understanding the variable importance will prove useful as a simple pr edictor of flexibility from an amino acid sequence. This will aid in better understanding of phenomenon underlying the average flexibility and thus, will pave a way for rational design of therapeutics.
Keywords: AGC kinase, Protein flexibility, Data mining, Classification and regression tree (CART), Bioinformatics
Every biological molecule is characterized and set apart from other biomolecules by a definite set of inherent intrinsic properties. Being the determinant of some vital functions like transport of metabolites (Anderson et al., 1990; Spurlino et al., 1991), catalysis (Bennett and Steitz, 1978; Remington et al., 1982) and regulation of protein activity (Perutz, 1970; Perutz, 1989) etc, average flexibility holds prime importance. Eukaryotic proteins demonstrate higher flexibility which influence conformational ability required in important biological processes like molecular recognition, interaction, assembly and modification. Moreover, protein flexibility is also known to influence stability and folding. There has been a sudden spur of interest in studies related to flexibility of proteins owing to discovery of role of some highly flexible proteins with implications in life threatening diseases like AIDS (HIV gp41) and scrapie (Chan et al., 1997). A comprehensive knowledge of fundamental nature of average flexibility will facilitate the unraveling of structurefunction relationship and will also aid in development of novel therapeutics (Teague, 2003).
AGC protein kinase family, one among the eight ePK families defined in the Kinbase, includes many important enzymes such as cyclic nucleotide and calcium-phospholipid dependent kinases, ribosomal S6-phosphorylating kinases, G protein-coupled kinases, and few others. The AGC serine threonine kinases, known for phosphorylating sites surrounded by basic amino acids, are involved in many intra–cellular signaling pathways, critical cellular processes and control cell growth, differentiation and cell survival. Their crucial role in transmembrane signaling process hints on the importance of features of AGC kinases which may be responsible for membrane localization (Peterson and Schreiber, 1999). This group of protein kinases shares similarity within the catalytic domain and is characterized by similar mechanism of activation. Deregulation of AGC kinases is known to have implications in several diseases like Cancer, Diabetes, neurodegeneration, and thus, AGC kinases represent several attractive targets for small inhibitors of therapeutic significance (Breitenlechner et al., 2003).
Their stringent spatio-temporal regulation is attained through loop phosphorylation and repositioning of the key catalytic and substrate binding regions which indicates the importance of flexibility in these proteins (Kannan et al., 2007). There is preponderance of literature on flexibility of proteins but elucidating the effect of parameters influencing it is cumbersome. This study aims at exploring the importance of different parameters influencing the average flexibility of AGC kinase family using data mining approach.
Sequence Collection and Pre-Processing
Protein sequences of the enzymes belonging to AGC family of protein kinase super family in FASTA format were collected from the non redundant (NR) protein database of NCBI (http://www.ncbi.nlm.nih.gov). Partial sequences were excluded from the study and sequences were again put to manual filtering so as to minimize the redundancy. This approach resulted in 600 sequences from the total 1259 sequences of AGC family available in the database were obtained. Out of these, sequences belong ing to Homo sapiens (59) and Mus musculus (56) were considered for this study.
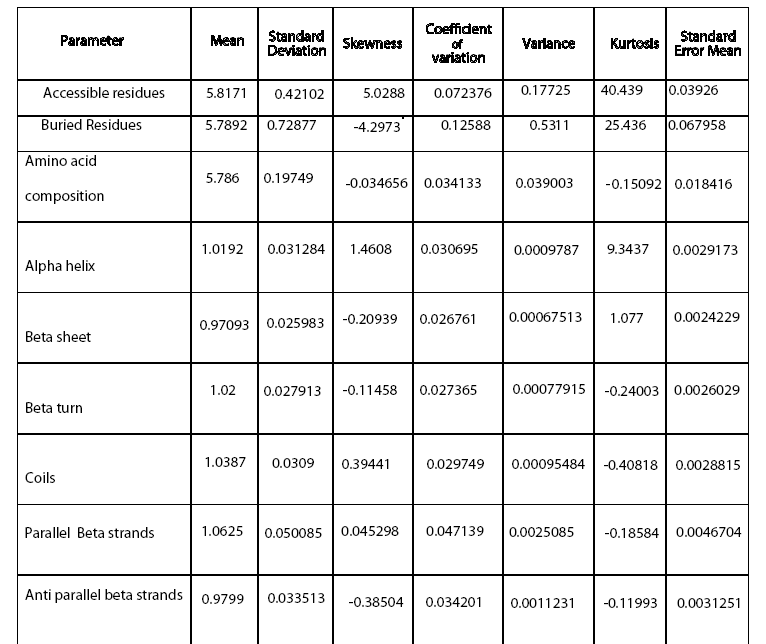
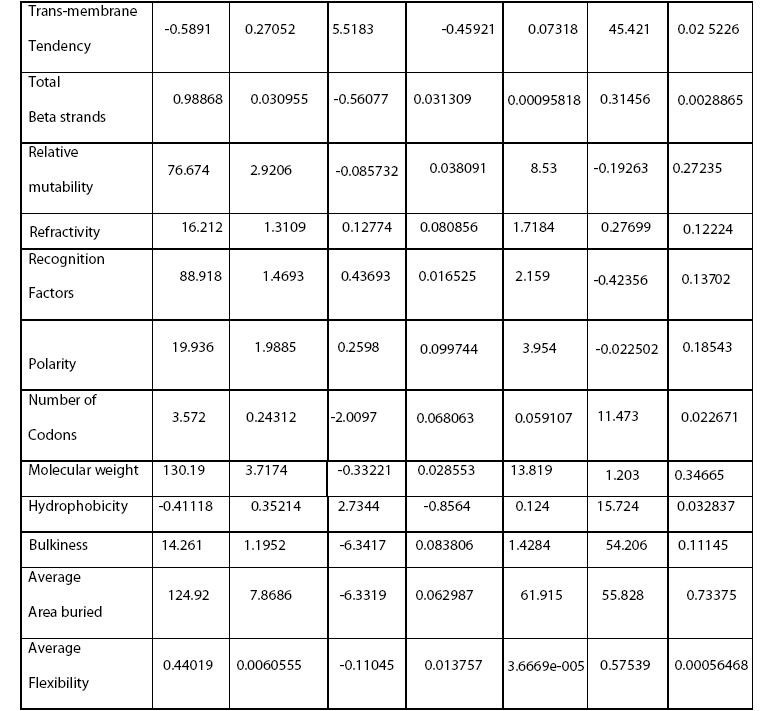
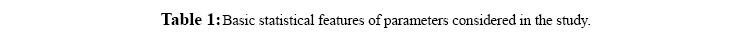

Figure 1: Frequency distribution chart for different parameters generated in CART 14 trees with different complexities and error values obtained using CART based on plitting criteria are reflected in table 2. Out of these trees, tree with 21 terminal nodes with minimum complexity and re-substitution relative error of 0.08501 and cross validated error of 0.72543 ± 0.12560 generated by Least Square splitting criteria was selected for generating decision rules. The topology of tree and error rate is represented in Figure 2. Splitters for the regression tree are shown in Figure 3. Decision rules obtained using CART are summarized in table 3 (Supplement).
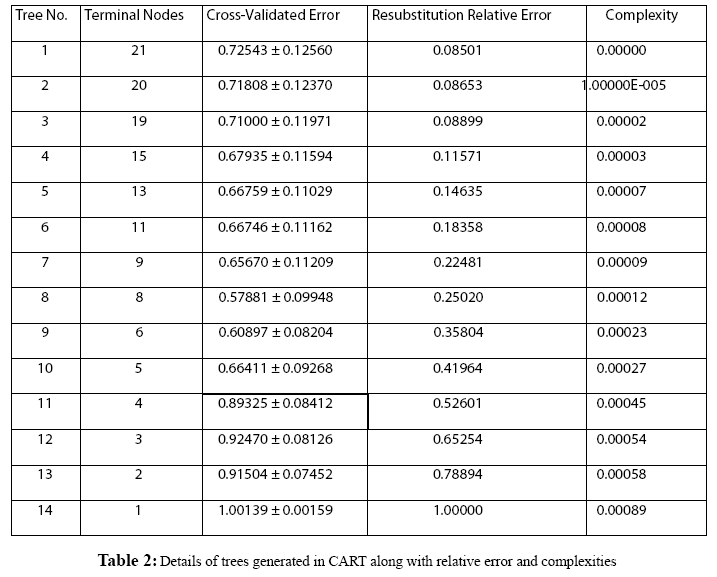
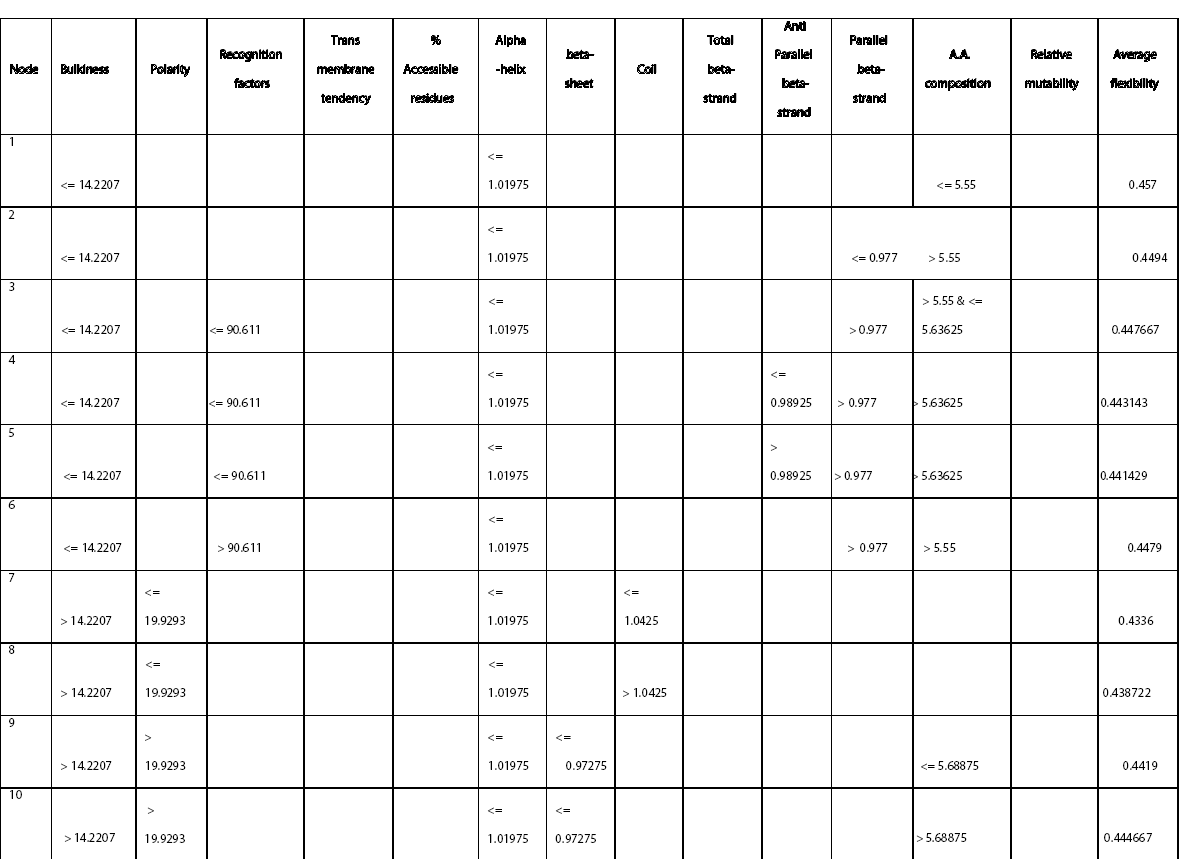
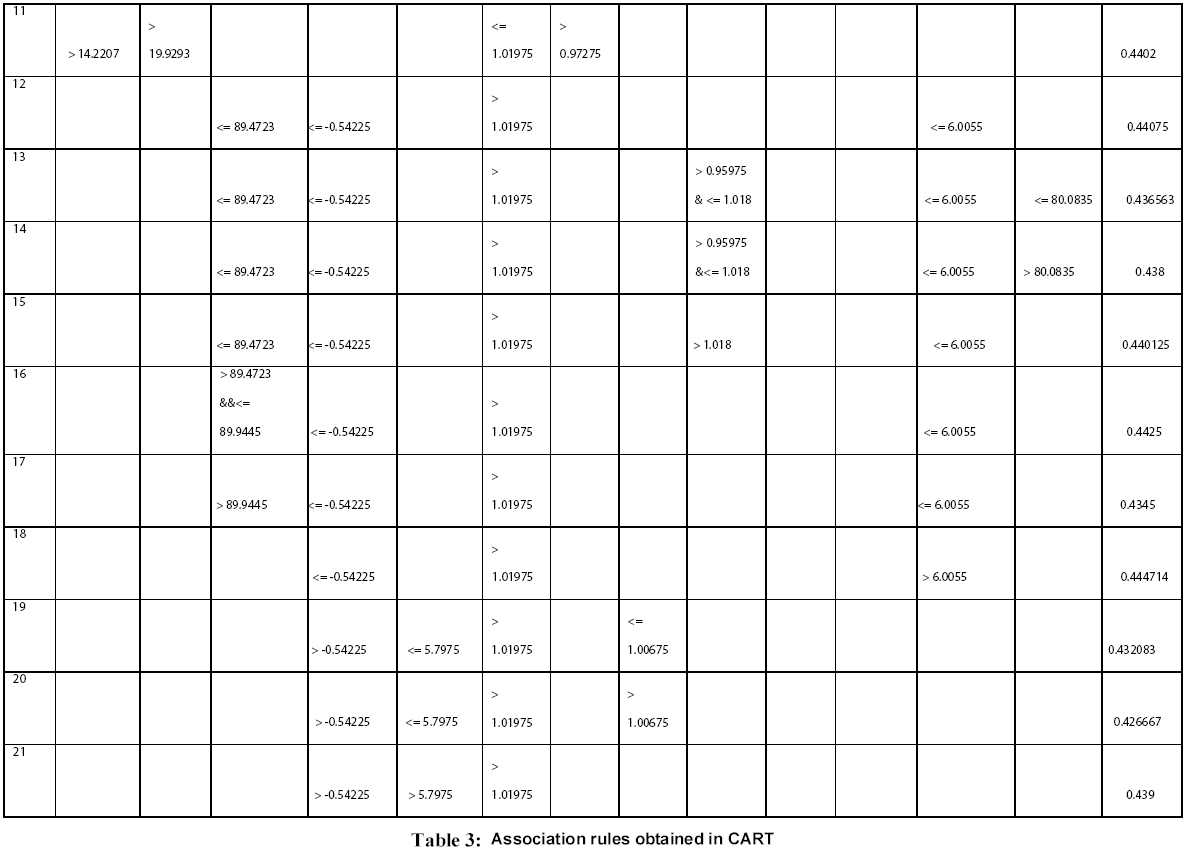

Figure 2: The tree sequence of lowest complexity which yielded 21 terminal nodes (A) with the cross validation error rate (B) and terminal node box plot(C).

Figure 3: Details of splitter for the Decision tree
Rules derived from CART can be interpreted in simple context of “If “and “Then” based statement and thus are self-explanatory. For example: Rule 1 can be interpreted as
Rule 1: IF “BULKINESS <= 14.2207” & “ALPHA -HELIX <= 1.01975” &” A.A COMPOSITION <= 5.55”, THEN “AVERAGE FLEXIBILITY=0.457”.
Rule 14: IF “RECOGNITION FACTORS<= 89.4723” &“TRANSMEMBRANE TENDENCY<= -54225” & “ALPHA -HELIX > 1.01975” & “TOTAL BETA-STRAND> 0.95975&<= 1.018” & “A.A. Composition<= 6.0055” & “RELATIVE MUTABILITY<= 80.0835”, THEN “AVERAGE FLEXIBILITY= 0.436563”
Variable Importance
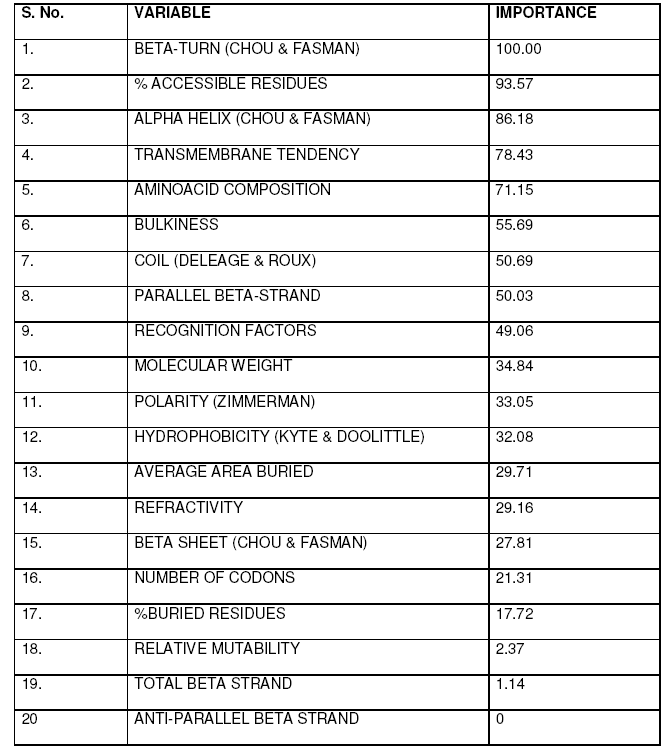
Importance of different variables was calculated based on predefined scores in CART and summarized in Table 4.
Dynamic nature of proteins, conferred by their structural flexibility, is associated with function. Average flexibility, an innate property of proteins is being recognized with implications in many important physiological processes recently (Wright and Dyson 1999; Bright et al., 2001; Dunker et al., 2001; Namba, 2001). Recognition of several highly flexibile proteins in some pathological conditions have led to the momentum in studies related to the flexibility of proteins. The huge gap in number of sequence and structures in PDB limits the utilization of 3-dimensional structure for deriving features affecting flexibility like Bfactors. In unavailability of such data, sequence composition and secondary structure provides a rough estimation of structural properties. This warrants the need for an alternate and simplistic approach for determining the effect of various parameters on average flexibility in an easy to understand quantitative relationship. Data mining approaches based on decision tree based methods have been successfully exploited in elucidating importance of features affecting important biological processes (Banerjee et al., 2007). CART has been exploited in microarray studies (Boulesteix et al., 2003), ecological studies (De’ath and Fabricius, 2000), risk prediction (Gottschalk et al., 1998), diseases diagnosis (Hermanek and Holzmann, 1994) and social studies (Özge et al., 2004).
The dataset comprising of various derived features was used to elucidate decision rules by CART that can serve as rule of thumb for finding the effect of different parameters on average flexibility, which is virtually impossible to calculate in a lab simultaneously using conventional approaches. Among the secondary structure features, beta turn, alpha helix, coil, parallel beta strand, beta sheet and total beta strands were found to influence the average flexibility in descending order. Among sequence features, % accessible residues, trans-membrane tendency, amino acid composition, bulkiness, recognition factors, molecular weight, polarity, hydrophobicity, average area buried, refractivity, no. of codons, % buried residues, and relative mutability were observed to affect the average flexibility in decreasing order(Table 4). Beta turns were found to have maximum impact while total beta strand were found to have minimum effect on average flexibility of the proteins considered in the study. As more and more studies are advocating the inclusion of protein flexibility in docking algorithms, it will be interesting to gain an insight on features influencing the flexibility of proteins. It is speculated that an extensive knowledge of protein flexibility and the various parameters contributing towards is important for rational drug design. Such an approach will lead to better understanding of underlying biological phenomena and aid in enzyme engineering processes.
Authors thank Dr. J.S.Yadav, Director, IICT for his continuous support and encouragement. We thank anonymous reviewers for their critical suggestions for the improvement of the manuscript.