Journal of Alcoholism & Drug Dependence
Open Access
ISSN: 2329-6488
ISSN: 2329-6488
Review Article - (2017) Volume 5, Issue 1
Alcohol has always been present in human life, and currently it is estimated that 50% of women of childbearing age consume alcohol. It has become increasingly clear over the last years that alcohol exposure during fetal development can have detrimental effects on various organ systems, and these effects are exerted by alcohol through multiple means, including effects on free radical formation, cellular apoptosis, as well as gene expression. Fetal alcohol exposure can lead to a spectrum of short term as well as long-term problems, with Fetal Alcohol Syndrome being on the more severe end of that spectrum. This syndrome is morbid, yet preventable, and is characterized by midfacial hypoplasia, thin upper lip, widely spaced small eyes, long smooth philtrum and inner epicanthal folds. Other findings include growth restriction as well as various neurodevelopmental abnormalities. This article is the first comprehensive review combining the molecular as well as the gross physiological and anatomical effects of alcohol exposure during pregnancy on various organ systems in the body. Our knowledge of these various mechanisms is crucial for our understanding of how alcohol exposure during fetal development can lead to its detrimental effects.
Keywords: Fetal alcohol exposure; Apoptosis; Cardiac defect; Pregnancy; Development; Alcohol and heart; Alcohol and brain; Reactive oxygen species
For thousands of years, alcohol has played a major part of human life. Since ancient times, various civilizations have used alcohol for leisure, trade, and religious rites. With the passage of time, multiple civilizations dwindled and vanished, whereas the use of alcohol only grew. In the present time, it is believed that as much as 40% of the world’s adult population consumes alcohol, with an average yearly alcohol consumption of 17.1 L per drinker [1]. Furthermore, it appears that the number of women, in particular, who consume alcohol, has been increasing, whereby it is estimated that 50% of women of child bearing age in the US consume alcohol in various degrees [2]. For nonpregnant women, physicians and many researchers define light drinking as 1.2 drinks per day, moderate drinking as 2.2 drinks per day and heavy drinking as 3.5 or more drinks per day [2]. Moreover, 40% of the 4 million annual pregnancies in the US drink alcohol, and among them 3-5% drink heavily [3]. It wasn’t until 1968 and 1973 that the first descriptions of a fetal syndrome linked to maternal alcohol abuse during pregnancy appeared in modern medical literature [4]. The current medical term for this syndrome is Fetal Alcohol Syndrome (FAS). It includes findings such as midfacial hypoplasia, thin upper lip, widely spaced small eyes, long smooth philtrum and inner epicanthal folds. Other findings include growth restriction as well as various neurodevelopmental abnormalities [2]. The incidence of FAS is estimated to vary between 0.5 and 3/1000 live births in the USA and Canada [5]. Multiple factors are involved in determining the severity of FAS in an afflicted individual. These include: Quantity of alcohol consumed during pregnancy, frequency of alcohol use, timing of maternal drinking in terms of gestational age, as well as multiple other maternal risk factors such as age, gravidity, nutrition, socioeconomic status, and metabolic and genetic influence [6,7]. Recent studies have also made clear that the pattern of drinking can also affect the severity of FAS, whereby binge drinking, defined as consuming >48 g of alcohol in one occasion [7], was found to produce very high maternal blood alcohol concentrations, thereby causing the most damage [6]. Given the detrimental effects of alcohol on fetal development, the American Academy of Pediatrics, as well as the American College of Obstetricians and Gynecologists both recommends abstinence during pregnancy for pre-conceptional and pregnant women [2].
Although chronic low alcohol levels have been shown to play a positive effect on cardiac structure and contractility in non-pregnant individuals [8]. This review article aims at covering the varying negative effects of alcohol in pregnancy, specifically on fetal development, based on timing of exposure during pregnancy, as well as the molecular and pathophysiological characteristics of Fetal Alcohol Exposure (FAE) in vital body organs such as the brain, heart and liver. Also, this article will shed light on the long term effects of FAE in the adult life of affected patients. Finally, this review article derives its importance from being the first article to combine the molecular as well as the gross physiological and anatomical effects of alcohol exposure during pregnancy on various organ systems in the body.
An extensive web search on PubMed was conducted for articles pertaining to the topic. The following are some of the search terms that were used: Fetal alcohol syndrome, fetal alcohol exposure, apoptosis, alcohol and heart, pregnancy, embryo development, reactive oxygen species. With few exceptions, all the articles were published after the year 2005. 150 articles were carefully read, and from those, information was cited from 73 articles.
It is known that the survival of the foetus during pregnancy depends on the exchange of nutrients and respiratory gases with the mother through a very vital gestational organ, the placenta. Along with many other functions, this organ is also known to acts as a barrier to multiple toxins and chemicals, hindering their transmission from maternal blood to fetal blood. However, ethanol can readily cross the placenta, subsequently making its way to fetal blood [9]. Furthermore, alcohol exposure time tends to be prolonged in the fetus due to its decreased capacity to Eliminate Ethanol (EtOH) secondary to low fetal alcohol dehydrogenase activity [10] and due to the reabsorption of EtOH that remains trapped in the amniotic fluid [9]. Studies have shown that even moderate amount of alcohol consumption can lead to FAS [9], partly due to this amplification system. Many studies in literature have attempted to understand. The mechanisms by which EtOH can affect fetal development. These mechanisms can be divided into 4 major categories: Molecular, genetic/epigenetic, metabolic, and cellular.
Ethanol (EtOH) has been found to cause apoptosis of neural crest cells [11], a group of progenitor cells that can give rise to a multitude of cell types including neurons, glial cells, and mesenchymal cells [12]. This ethanol induced apoptosis has been shown to be mediated through different molecular mechanisms, one of which is increased mRNA expression of the Siah1 protein, a member of the E3 ubiquitin ligase family [11], which triggers apoptosis through phosphorylation of p38 MAPK, leading to activation of the p53 signaling pathway. p53 is known to be a transcription factor for many pro-apoptotic genes [13]. Furthermore, ethanol has been found to be involved in other apoptotic signaling pathways, including Fas/Fas-L [14], Bcl-2 family [15], and caspase signaling systems [16]. However, these mechanisms have not been clearly elucidated [13].
In addition to its role on apoptosis, EtOH has been found to affect cellular respiration in rat cardiac cells by influencing the mitochondrial membrane, specifically Cardiolipin, a major glycerol-phospholipid required for mitochondrial enzymes involved in energy metabolism within the cell [17]. This latter mechanism has been explained by the fact that metabolites of alcohol, specifically fatty acid ethyl esters, decrease the activity of monolysocardiolipin acyltransferase (MLCL AT), which is the rate-limiting enzyme in the transformation of monolysocardiolipin to Cardiolipin in cardiac mitochondria [17]. In fact, Taylor et al., found that the activity of the MLCT AT enzyme was 36% lower in newborn rats with in-utero exposure to alcohol [10]. Also, not only do fatty acid ethyl esters decrease the activity of MLCL AT, but they also seem to play a role in altering protein expression and uncoupling oxidative phosphorylation by directly binding to mitochondria in intact cells [18]. Furthermore, a third mechanism is alcohol mediated oxidative damage. An example of this is the increased production of Reactive Oxygen Species (ROS) in the brain by catalase mediated acetaldehyde production from alcohol, thus leading to impairment of the blood brain barrier and neurodegeneration in the developing fetus [19-21]. A fourth major mechanism by which EtOH has been found to affect fetal development, apart from apoptosis, oxidative stress, and cellular respiration, is acidemia mediated decrease in key amino acid concentrations, especially that of Glutamine which plays a role of paramount importance in fetal growth, as well as synthesis of nucleotides [22], the antioxidant glutathione [23], and the brain neurotransmitter glutamate [23]. This alcohol mediated reduction in glutamine and other related amino acids in the plasma were found to be around 21-30% in exposed pregnant Ewes as per a study done by Washburn et al. [24]. It has been found that blood pH decreases proportionally to increases in ethanol blood concentration since ethanol consumption leads to a mixed respiratory and metabolic acidosis [25]. This acidosis leads to increased glutamine uptake and metabolism by renal mitochondria [26]. Acidosis also increases renal expression of N-glutamine transporter (SNAT 3), which increases glutamine uptake by the kidney [27]. When it comes to binge drinking, Ramadoss et al. [28] created a Ewe model mimicking third trimester binge drinking, and showed acidosis mediated drop in plasma glutamine and other amino acid levels including threonine, serine, glutamine, glycine, alanine and methionine [29]. This drop in amino acids directly affects the synthesis of various functional and structural proteins (Figure 1).

Figure 1: Effect of alcohol exposure on fetus. MLCT AT: Monolysocardiolipin acyltransferase; BBB: Blood Brain Barrier; ROS: Reactive Oxidant Species; Legend 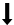 : Decreases;
: Decreases; 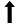 Increases; → leads to;
Increases; → leads to; 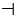 inhibits. Explanation: Alcohol consumption in pregnancy leads to: 1) Decrease in MLCT AT which will lead to decrease in cardiolipin ( a major glycerophospholipid required for mitochondrial enzymes involved in energy metabolism; 2) Acedemia which will decrease glutamine which is important for fetal growth and nucleotides synthesis; 3) Increase in Siah1 protein leading to phosphorylation of p38MAPK leading to increase activity of p 53 and eventually apoptosis; 4) Increase in ROS which will: a) impairment of BBB leading to Neurodegeneration of fetus; b) increase in NF-kB which will inhibit the increase of ROS through a protective mechanism (autophagy).
inhibits. Explanation: Alcohol consumption in pregnancy leads to: 1) Decrease in MLCT AT which will lead to decrease in cardiolipin ( a major glycerophospholipid required for mitochondrial enzymes involved in energy metabolism; 2) Acedemia which will decrease glutamine which is important for fetal growth and nucleotides synthesis; 3) Increase in Siah1 protein leading to phosphorylation of p38MAPK leading to increase activity of p 53 and eventually apoptosis; 4) Increase in ROS which will: a) impairment of BBB leading to Neurodegeneration of fetus; b) increase in NF-kB which will inhibit the increase of ROS through a protective mechanism (autophagy).
Concerning the genetic aspect, it is known that the relationship between alcohol and genetics goes both ways: On one hand, an individual’s genetic makeup can have a direct effect on alcohol metabolism and hence alcohol blood concentration level. Various enzymes are involved in the metabolism of alcohol, such as alcohol dehydrogenase, aldehyde dehydrogenase, and CYP2E1 [30]. Different variations of the genes encoding for these enzymes in the mother and fetus can lead to differences in enzyme activity and hence differences in alcohol metabolism and alcohol related damage [30]. On the other hand, alcohol has been shown to affect gene expression without having direct effects on the gene sequence itself [31]. This is referred to as epigenetic processes, including DNA methylation, histone modification and RNA interactions. Starting with DNA methylation, it is the process of transferring a methyl group by DNA methyltransferase to one of the four DNA nucleotides, thereby leading to a condensed chromatin formation, thus effectively silencing gene expression [32]. It is folate and B12, and their involvement in the formation of SAM (s-adenosyl methionine), that are involved in the supply of these methyl groups [33]. Alcohol excess is known to affect folate metabolism and availability, subsequently influencing DNA methylation, affecting fetal growth and development [34]. Furthermore, alcohol’s effect on folate levels also impacts histone activity, as folate is known to be involved in histone methylation [34]. Histones are principle structural proteins of chromosomes in eukaryotes, and certain histone types are involved in the formation of DNA-histone units called nucleosomes [34]. The interaction of these nucleosomes with other histone types is what determines the transcriptional activity of chromatin. Alcohol has been shown to lead to hypermethylation of Lysine 4 in histone 3 and hypomethylation of Lysine 4 in histone 9 in studies done on hepatocytes [34]. It is suspected that these distinct methylation changes in histones can affect chromatin structure, and thus gene expression level [34]. Other alcohol related changes in histone include histone acetylation, a process that has epigenetic effects on various organs. It was demonstrated that histone acetylation in lung cells leads to increased apoptosis [35]. In the amygdala, histone acetylation was shown to change gene expression of Neuropeptide Y, an important molecule in the autonomic nervous system [36]. Also, in the heart, an experiment done by Pan et al. demonstrated that H3 hyperacetylation could be linked to structural heart defects such as biventricular enlargement and septal thinning [37,38]. Zong et al. (2010) similarly found that both low and high levels of alcohol lead to hyperacetylation of histones; but interestingly, it is only at high levels that genes involved in cardiac structure such as GATA 4, Mef2C, Tbx5 are affected [38].
Given the various mechanisms by which alcohol induces damage to cellular development and function, the body has devised various protective mechanisms to hinder the damaging effects of alcohol. One such mechanism is neuronal cell autophagy, whereby ethanol-induced ROS (mainly originating from NADPH oxidase) regulate the activities of certain transcriptional factors such as NFKB, TP53 and NFE2L2. These factors affect various signaling pathways involved in autophagy and apoptosis of neuronal cells, thereby blocking ROS induced neurodegeneration [39]. Chen et al. touched upon one of the autophagy signaling pathways, reporting that ethanol enhances autophagy through inhibition of the mTOR (Mammalian Target of Rapamycin) pathway [40]. Furthermore, it was found that ethanol leads to a moderate increase in the expression of Nrf2, a main regulator of expression of antioxidant genes. It is important to note that it has been shown that this moderate increase in Nrf2 in relation to ethanol is only an adaptive response, and that this response is not enough to prevent ethanol induced apoptosis [41]. However, understanding these protective mechanisms against alcohol is vital, since it sheds light on possible treatments that can make use of these mechanisms to hinder the negative effects of alcohol.
Finally, not only does ethanol damage cells by various mechanisms, but it is suspected to also have varying effects, depending on the gestational age when ethanol exposure occurs. This has been studied in literature at the level of neuronal cells. It has been shown that certain regions such as the ventromedial forebrain and mesodermal cells are affected when EtOH exposure occurs in the first trimester. On the other hand, third trimester exposure to EtOH seems to affect different brain regions such as the prefrontal cortex, hippocampus, cerebellum as well as the corpus callosum [42,43].
After reviewing the various major mechanisms by which EtOH mediates its detrimental effects on fetal development, we will now proceed with the discussion of the various physiological and anatomical effects of alcohol on different organ systems, specifically on organs affected most by alcohol exposure such as the liver, the heart, and the brain.
Beginning with the liver, earlier studies have shown that children with fetal alcohol syndrome may have abnormal liver function tests, along with fibrosis and fatty hepatic degeneration [44]. Furthermore, it was demonstrated increased fat deposits in hepatic cells of rat fetuses when they were exposed to alcohol in late trimesters [45]. However, more recent studies on sheep by Sozo et al. have failed to show any change in fetal liver morphology [46]. The effect of alcohol in these studies was found to affect iron homeostasis with change in gene expression of hepcidin and ferroportin, and subsequent drop in fetal liver ferric content when alcohol exposure occurred in the third trimester [46]. A prospective study by Carter et al. also demonstrated the effect of alcohol on iron homeostasis, whereby there was a 3.6 times increased risk of iron deficiency anemia in infants of mothers who binge drank during pregnancy. This was explained by possible disruption of iron transport to the fetus via the placenta, in addition to the disruption of the absorption and storage of iron by the fetus. Interestingly, it was observed that alcohol exposed infants with iron deficiency anemia had delayed growth, and had a lag in weight gain and head circumference during the first year of life, and these delays could not be corrected after replacing iron stores in the newborn, suggesting that iron could be one of the instigating factors. However, sustainable effects of alcohol on infant growth seem to be driven to a greater extent through mechanisms other than iron deficiency [47].
In the brain, alcohol has been found to have both structural and functional effects on neural development. These include decreased brain volumes, defects in development of certain brain structures like the brainstem, cerebellum, corpus callosum, frontal lobe, and thalamic nuclei, as well as errors in the migration of neurons [48,49]. Radial glial cells, which are precursors for various cells within the nervous system such as neurons, astrocytes, and oligodendrocytes, are disrupted when exposed to alcohol [50]. Prenatal alcohol exposure has also been shown to reduce connectivity between the two hemispheres of the brain, notably in the para-central cortical regions, thereby delaying the maturation of brain networks, while simultaneously increasing the connectivity between somatosensory, motor, and brainstem networks [51,52]. Burke et al. demonstrated that alcohol exposure in monkeys during the third trimester led to a decrease in the number of neural progenitor cells in the olfactory bulb and dentate gyrus [53]. Cortical development has also been shown to be affected by alcohol exposure during fetal development by altering the migration of Gamma-Aminobutyric Acid (GABA) secreting interneurons that mediate the inhibitory/excitatory balance within the intracoritcal circuit [54]. Furthermore, children who have been exposed to alcohol in-utero were found to have difficulties in executive functioning, including difficulties in organizing thoughts, maintaining emotional stability, planning, and self-monitoring [49]. Motor impairment has also been described, whereby affected children were noted to have delays in fine motor skills, hand-eye coordination, and motor reaction time to visual stimuli [55,56]. Attention and learning capacity have also been affected by alcohol exposure in utero, and interestingly FAE children have been known to be misdiagnosed with Attention Deficit Hyperactivity Disorder (ADHD) due to their irritable behaviors and difficulty in maintaining attention [57]. Finally, the effects of prenatal alcohol exposure also include problems with social interactions and overall social skills, including hyperactive behaviors, communication difficulties, and problems with assuming responsibility, with more severe social impairment seen in females as compared to males [58].
As for the heart, it is important to note that this complex organ has multiple components, each originating from different precursor cells [59]. These components include cardiomyocytes, connective tissues, conduction system cells, as well as smooth muscle cells and endothelial cells [59]. The three sources of cardiac cell precursors are the cardiogenic mesoderm cells, the proepicardium, and the cardiac neural crest cells [59]. The latter specifically gives rise to the autonomic nervous system of the heart, along with playing a crucial role in heart septation and valvular development [59]. The importance of neural crest cells has been demonstrated in studies that showed a reduced great vessel diameter and atrioventricular valve leaflet volumes in embryos where these precursor cells were ablated. Interestingly, these heart defects were found to be similar to those seen in avian models of fetal alcohol syndrome, thus proposing that one possible mechanism for alcohol induced cardiac damage is through abnormal development of neural crest cells [60]. A possible explanation for the link between alcohol exposure and neural crest cell dysfunction could be that these cells tend to have higher sensitivity to free radicals, and thus higher rates of apoptosis, due to an inherent decreased endogenous superoxide dismutase activity [61-63]. Other studies have also proven that alcohol exposure can hinder neural crest cell migration [63]. Furthermore, studies performed on zebra fish have demonstrated that differences in the length of ethanol exposure can lead to different degrees of cardiac abnormalities during fetal development, whereby chronic alcohol exposure was more likely to lead to significantly more severe endocardial cushion defects as compared to defects seen in short term alcohol exposure. In fact, hearts exposed to alcohol chronically during development were found to be small with no internal tissue separating the chambers, due to the elimination of endocardial cushion cells [64]. Apart from the role of alcohol exposure length, avian models have also demonstrated the role of ethanol concentration in determining degree of cardiac effects, whereby valvular abnormalities were found in 68% of hearts from embryos exposed to high concentrations of alcohol [65]. Similarly, studies performed on larvae showed that exposure to higher concentrations of alcohol led to drastic dorsal aorta destruction and segmental artery coarctation. Also, hearts of the high alcohol concentration exposure group showed morphological differences and were significantly smaller than the control group [66]. However, even in low alcohol concentrations, the heart rates of the exposed hearts were found to be slower than the control hearts, with this decrease in heart rate noticed to be more pronounced in hearts exposed to higher concentrations [67].
After discussing the effects of alcohol exposure on various organ systems during fetal development, it is important to shed light on more lasting effects of in utero alcohol exposure. In fact, multiple studies have demonstrated that there are long-term consequences of fetal alcohol exposure, which has been described by the Barker Hypothesis. Barker suggested that certain patterns of fetal growth could affect various body functions including blood pressure control, sensitivity to insulin, metabolism of glucose, as well as cardiac functions in adult life. Disruptions to these patterns of growth by factors like maternal malnutrition, stress, and alcohol exposure can affect these aforementioned physiological functions on the long term. This hypothesis has also been termed the “Developmental Origins of Adult Health and Disease” or “DOHaD” [68-71]. Furthermore, studies have shown that early life ethanol exposure can affect mesenchymal stem cells, the main cells responsible for tissue repair, thus increasing susceptibility to disease later on in life. Leu et al. found that mesenchymal stem cells of rats exposed to ethanol during the third trimester equivalent were resistant to osteogenic and adipogenic inductions in comparison to controls. In these experiments, it was found that expression of multiple proteins like osteocalcin and alkaline phosphatase was impaired [72]. Other experiments on mouse models have shown consequences like reduced weight, survival, and immune response, specifically B and T cell response, in alcohol-exposed mice. Furthermore, there were an increased number of tumors in those mice as compared to controls. Other observed effects included increased insulin resistance, hypertriglyceridemia, as well as increased chronic inflammatory processes such as arthritis [73]. Given these devastating effects, we believe that it is crucial that fetal alcohol exposure is identified early on by obstetrician and gynecologists as well as pediatricians/neonatologist, in order to actively manage and prevent the long-term sequelae.
Alcohol use has been steadily increasing, and its consumption by women continues to be on the rise. All evidence shows that alcohol plays a detrimental effect on fetal development. As discussed extensively in this article, these effects span multiple organ systems from the heart and brain to the liver, kidneys, as well as other major organs. Alcohol exerts these effects in multiple ways, with influences on free radical formation, cell programming and cell death, as well as gene expression, among many other mechanisms. The exposed body reacts by certain adaptive responses, such as an increase in Nrf2, which affects the expression of anti-oxidant genes, but this increase is not sufficient to prevent ethanol induced apoptosis. It is also clear that the harmful effects of alcohol vary in relation to the pattern of drinking, with more harm done with binge drinking than with chronic alcohol use, as well as to the level of alcohol exposure during pregnancy. Furthermore, there is a temporal component to alcohol-mediated damage, whereby exposure at a specific gestational age during the pregnancy can lead to a variety in both nature and severity of the effect. The information presented in this article is crucial for our understanding of the development as well as the prevention of the effects of fetal alcohol exposure, which are morbid, yet preventable, and affecting thousands of individuals worldwide, and leading to chronic medical, developmental, and social problems in affected patients. Effort needs to be done in order to increase awareness about the harmful effects of even minimal amounts of alcohol consumption during pregnancy. Women need to be counseled to stop alcohol intake completely during their preconception visits, and the question of whether or not they are consuming alcohol while pregnant needs to be brought up during every prenatal follow up visit with the obstetrician. Furthermore, more research is needed to uncover hidden pathways through which alcohol can affect cell structure and function. Understanding these different pathways and attempting to block them may eventually allow us to mitigate the harmful effects of alcohol exposure during fetal development. It is important to note that many cases of fetal alcohol exposure are discovered later in life, sometimes even during adulthood. This is due to the epigenetic effects of alcohol, leading sometimes to late manifestations of abnormal development. Therefore, more effort needs to be made in order to discover fetal alcohol exposure as early as possible and subsequently managing the chronic effects early on and attempt to decrease the morbidity. Currently, it is estimated that 40000 children in the U.S. are born with FASD every year. This figure represents 2-5% of school-aged children. This is preventable mainly through education for parents, and through raising awareness of the harmful effects of alcohol during pregnancy. Also, training and guidance could be provided for medical professionals in the field of women’s health to properly council women of childbearing age. Schools also play a role by creating special needs programs so that affected children are capable of reaching their full educational potential [74]. Finally, we conclude by saying that alcohol consumption during pregnancy has a toll, shared by both affected individuals as well as the society in which they live, given the various socioeconomic effects that Fetal Alcohol Exposure has. Therefore, it requires a collective effort in order to avoid paying this toll and to ensure the health of the future generations to come.
This work was supported in part by grants 1 R15 AA019816-01A1 and 2G12 RR003048 RCMI, Division of Research Infrastructure to GEH; Howard University College of Medicine.