Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Editorial - (2014) Volume 2, Issue 1
With the emergence of several multi-drug resistant bacterial strains the search for novel antibiotics has taken a renewed urgency [1,2]. To date, a class of natural products called non-ribosomal peptides (NRPs) has been successfully used as antibiotics [3-5]: penicillin, cephalosporin, bacitracin, rifamycin, erythromycin, vancomycin and more recently, daptomycin, which is used to treat Methicillin-resistant Staphylococcus aureus (MRSA) infections [6]. NRPs are secondary metabolites produced by bacteria and fungi to compete with each other and infect their hosts. Hence it is ironic that to find inspiration for novel antibiotics we look to bacteria to see how they kill other strains of bacteria. Nevertheless, the track records of NRP use in pharmaceutical applications speak for themselves, not just as antibiotics but also as other drugs: bleomycin as an anticancer agent, cyclosporine A or rapamycin as immunosuppressants, etc [3,4]. In addition to serving as therapeutic agents NRPs are also etiological agents in microbial infections [4]. Hence NRPS studies will not only allow us to understand biosynthetic techniques of producing current and hopefully novel antibiotics but will also be crucial in understanding bacterial infections.
Despite their fascinating chemical diversity NRPs are synthesized in bacteria and fungi with a functionally conserved modular organization of multi-domain enzymatic systems, called non-ribosomal peptide synthetases (NRPSs) [3]. NRPSs function in an assembly line-like manner with each module adding a single substrate during NRP chain elongation. Such modular organization and assembly line-like synthesis presents an appealing idea for the biocombinatorial approach to generate novel antibiotics. Swapping of modules and/or domains amongst different NRPS systems, so called NRPS reprogramming, may produce novel compounds with antimicrobial activity [7,8]. But is such NRPS reprogramming a viable option for new antibiotics?
NRP chemical diversity is dictated by the arrangement of the catalytic units in their respective NRPSs. A canonical module consists of three key domains (Figure 1) [3,9]. A thiolation domain (T) posttranslationally modified with a 4’-phosphopantetheinyl (Ppant) arm carries the elongating NRP. An adenylation domain (A) selects, activates and loads the substrate on the Ppant arm with a thioester linkage. Finally, condensation domains (C) catalyze peptide bond formation between the substrates loaded on T domains of sequential modules. Condensation domains are sometimes replaced by cyclization domains (Cy) that carry out both condensation and heterocyclization (cyclodehydration of serines or threonines to oxazolines, cysteines to thiazolines) [3]. Domain organization within a module generally follows CAT architecture where the individual domains may be in cis or in trans. The last module has a thioesterase domain to release the final product, which at times is macrocyclized (e.g. enterobactin) before release. Additionally, there are other tailoring domains that act in cis or in trans: epimerization domains which racemize the peptide bond in the growing NRP (e.g. Yersiniabactin), oxidase domains that oxidize labile dihydroheterocycles to stable heterocycles (e.g. bleomycin), methyl transferases that introduce a methyl moiety (e.g. epothilone), halogenases that add a chloride (e.g. coronamic acid), to name a few [3].
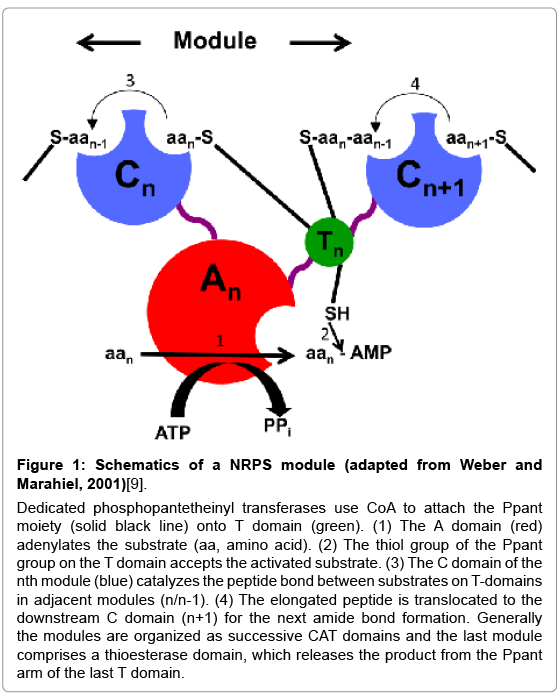
Figure 1: Schematics of a NRPS module (adapted from Weber and Marahiel, 2001)[9].
Dedicated phosphopantetheinyl transferases use CoA to attach the Ppant moiety (solid black line) onto T domain (green). (1) The A domain (red) adenylates the substrate (aa, amino acid). (2) The thiol group of the Ppant group on the T domain accepts the activated substrate. (3) The C domain of the nth module (blue) catalyzes the peptide bond between substrates on T-domains in adjacent modules (n/n-1). (4) The elongated peptide is translocated to the downstream C domain (n+1) for the next amide bond formation. Generally the modules are organized as successive CAT domains and the last module comprises a thioesterase domain, which releases the product from the Ppant arm of the last T domain.
In theory, the NRPS reprogramming idea is deceptively simple; create a diverse library of bioactive compounds based on a successful antibiotic chemical scaffold from a (hopefully) one-pot reaction of NRPS enzymes and substrates. This diverse library of compounds may then be checked for viability as antibiotics as well as bioavailability. The idea has credibility since domain boundaries in the mega-enzymatic systems are moderately easy to identify from protein sequence alignments. Hence, hypothetically, one should be able to add modules from different NRPSs (in cis or in trans) to produce a new NRPS system that can generate at least one new compound, if not a library. However, the growing NRP chain is covalently harbored on T domains (via Ppant arms) and hence the T domain must interact with at least two other domains to elongate the chain (A & C). Furthermore the C domain has to recognize two T domains, one cognate and a non-cognate T domain from a different NRPS module. And herein lies the problem. The NRP chain growth is guided by multiple interactions and hence domains must adopt multiple conformations to guide NRP chain elongation. For A and C domains the active site may still be modified to carry out loading and condensation reaction with the newly designed NRPS system. But for T domains the conformers necessary to interact with a non-cognate C domain might not be present. Biochemical studies have demonstrated selectivity for both substrate-domain and domain-domain interactions in NRPS systems [10]. Multi-domain structures solved by X-ray crystallography revealed only non-functional inter-domain orientations [11,12], indicating that NRPS domains must rearrange their orientations during NRP synthesis. This was also highlighted by solution Nuclear Magnetic Resonance (NMR) studies of a di-domain [13], where transient interactions were observed, demonstrating that NRPSs are not rigid multi-domain assemblies but are instead subject to transient, sequential domain interactions. Furthermore, multiple conformers in equilibria were observed for excised NRPS domains [14,15], hinting that domain interactions and product hand-off between domains may be propelled by conformational selection. In short, the mechanism employed by cyclization domains and their interactions with carrier domains are poorly understood.
The dearth of knowledge regarding sequential domain interactions in NRPS arises from the lack of solution structures by NMR of various key domains. Solution NMR techniques are well suited to probe such transient interactions. Unfortunately the large size of C and A domains make NMR solution structure determination and interaction studies cumbersome. Indeed most structures of C and A domains are X-ray crystal structures and there has been limited success with identifying domain interactions. Nevertheless, NMR remains a promising tool of choice to study NRPS domain and inter-domain interactions [13]. In an ideal world if solution structures of all NRPS domains (at least A & C) were available and surface interactions between A & T, and C and it’s cognate T domains could be mapped at a molecular level, one could envision a higher rate of success in NRPS reprogramming. Mapping of such interaction surfaces might allow researchers to graft the sequence of the interaction interface on NRPS domains rather than a simple primary sequence exchanges found by sequence alignment. That is not to say that current research has had no success with NRPS reprogramming. However such studies are mostly limited to mutating A domain active sites to load non-cognate substrates on T domains [16-18]. In conclusion, the goal of NRPS reprogramming is still viable to generate novel antibiotics but more information about interaction interfaces has to be obtained for its ultimate success.s