Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Research Article - (2018) Volume 7, Issue 1
Background: The GlcNAc related disaccharides were reported to show inhibitory activity on breast tumor cells (MDAMB- 231). However, synthesis of disaccharides is lengthy, laborious and time consuming, and it is necessary to find more efficient methods to generate the disaccharide libraries. Random glycosylation has been an established practice to generate β-linked disaccharides library. Synthesis of α-linked disaccharide libraries by random glycosylation hasn’t been explored yet. We present our results on the synthesis of α-linked GlcNAc related disaccharide libraries by random glycosylation in this article.
Results: Employing random glycosylation as a key step on an unprotected GlcNAc monosaccharide, and after deprotection and careful purification, few α-linked GlcNAc related disaccharide libraries were obtained in acceptable ratios, and the results were confirmed by mass and NMR spectral data.
Conclusion: Random glycosylation was demonstrated as an efficient method for the synthesis of α-linked GlcNAc related oligosaccharide libraries.
Keywords: Oligosaccharide; Random glycosylation; Combinatorial chemistry; α-Linked GlcNAc oligosaccharide library
Interaction between cell surface carbohydrate ligands and various protein receptors forms the basis of recognition events, which are fundamental to vastly diverse range of biological and pathological processes [1]. Cell surface carbohydrates constitute efficient receptors for hormones, toxins, bacteria and viruses [2]. Therefore, synthetic oligosaccharides can be screened as cell adhesion inhibitors or antigens from which specific antibodies can be triggered. However, oligosaccharides are very complex and diverse, and their synthesis is both lengthy and expensive [3]. Recent research showed that the most important residue in the structures of oligosaccharide epitopes recognized by the antibodies are the terminal fragments which normally are disaccharides, trisaccharides or tetrasaccharides [4]. This fact elicited lot of interest to synthesize a diverse array of small size (di, tri and tetra) oligosaccharide libraries for studying their biological properties instead of synthesizing the whole size oligosaccharides.
Specific disaccharides have been identified as markers for certain types of tumors. For example, the chondroitin sulfate disaccharides were reported to show inhibitory activity on breast tumor cells (MDAMB- 231) [5], 16 chondroitin sulfate disaccharides were tested on the more aggressive MDA-MB-231 cell line, a statistically significant decrease in cell viability was observed for some of disaccharides at 100 μg/mL concentration, and the results indicated that both the number and position of the sulfate groups present in the chondroitin sulfate disaccharide have an effect on MDA-MB-231 cell viability. These results stimulated lot of interest among chemists to find the more efficient methods to synthesize the small sulfated oligosaccharide libraries.
Accordingly, using an orthogonal protection strategy, sulfated Gal- β1,3/4-GlcNAc disaccharide library was reported by Tu et al. [6] while synthesis of 48 disaccharide building blocks for assembling of a heparin oligosaccharide library was achieved by Hung research group [7]. Further, from a common intermediate possessing orthogonally removable protective groups, all 16 disaccharide sulfates were systematically synthesized by Suda et al. [8]. These syntheses are both lengthy and time consuming, and there is a large scope to find more efficient methods for generating the oligosaccharide libraries.
Random glycosylation on unprotected glycosylation acceptors can generate oligosaccharide libraries containing all possible oligosaccharides with certain configuration [9] Random galactosylation on an unprotected GlcNAc acceptor generated all sixpossible disaccharide library with α and β linkages, and six disaccharides were chemically synthesized to compare with the library in HPLC to confirm the results [10]. The ratio of six disaccharides is not good enough for their biological activity evaluation, even many reaction conditions and glycosylation donors were used in order to improve the ratio of six disaccharides, but none of them worked. However, random glycosylation of glucosamine and galactosamine related donors on unprotected GlcNAc acceptors gave the β-linked GalNAc and GlcNAc related disaccharide libraries with excellent ratios [11]. The α-linked disaccharide and trisaccharide derivatives may have altogether different interesting activities when compared to their β- linked counterparts, and not much has been reported on the synthesis of α-linked disaccharide libraries thus far.
In continuation of our interest in improving methods for random glycosylation to obtain biologically relevant oligosaccharides, we like to report herein the synthesis of galactose, glucose and mannose related α-linked disaccharide and trisaccharide libraries which were confirmed through NMR and MS spectral study. The target libraries are listed in Figure 1.
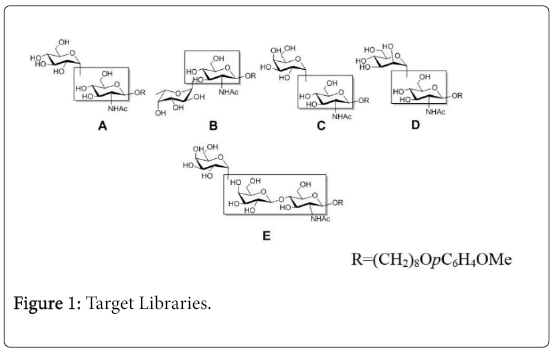
Figure 1: Target Libraries.
TLC was performed on silica gel 60-F254 and charred with H2SO4. 1H NMR spectra were recorded at 300 MHz (Bruker AM-300), 360 MHz (Bruker AM-360), or 500 MHz (Varian UNITY 500) on solution in CDCl3 (internal standard TMS, δ0), or D2O (internal standard acetone, δ2.23). MALDI MS was obtained from Kratos Kompact MALDI. Methanol was distilled from Mg, and toluene was distilled from CaH2.
Random glycosylation of glycosyl trichloroacetimidates (2-4) on GlcNAc 1
The mixture of 8- p-methoxyphenoxy-octyl N-acetyl 2-deoxy-2- amino β-D-glucopyranoside (1) (300 mg, 0.65 mM), glycosyl trichloroacetimidates (2, 3 or 4) (1.3 mM) and CaSO4 (500 mg) in dry dioxane (5 mL) were stirred at room temperature under argon for 30 min., then BF3•Et2O (29 mg, 0.13 mM) was added. After stirring at room temperature for another 14 hours, three drops of triethylamine were added to quench the reaction, and the reaction mixture was filtered through celite to remove the solid, washed with dioxane, evaporated, purified on a silica gel column eluted with dichloromethane and methanol (10:1), each fraction was checked by LC-MS, the unreacted acceptor, the donor hydrolysis products, and trace amounts of trisaccharides were removed, and the disaccharides were combined and hydrogenated in methanol in the presence of Pd/C for 48 hours at room temperature. After work up, the residue was purified on C-18 column with water and methanol to get the random glycosylation products as disaccharide libraries A, B and C. Spectral data for library A: 1H NMR (D2O, 500 Hz): δ5.170 (d, J=4.8 Hz), 5.142 (d, J=4.7 Hz), 4.846 (d, J=4.9 Hz); ESIMS: m/z 618 [M+H]+. Spectral data for library B: 1H NMR (D2O, 500 Hz): δ4.963 (d, J=4.8 Hz), 4.870 (d, J<4.0 Hz), 4.815 (d, J=4.7 Hz); ESIMS: m/z 602 [M+H]+. Spectral data for library C: δ5.220 (d, J=4.8 Hz), 5.154 (d, J=4.7 Hz), 4.873 (d, J=4.7 Hz); ESIMS: m/z 618 [M+H]+.
Random mannosylation on GlcNAc 1
8-p-methoxyphenoxy-octyl N-acetyl 2-deoxy-2-amino β-Dglucopyranoside (1) (260 mg, 0.62 mM) was dissolved into dry dioxane (2 mL), and per-O-acetyl mannopyranosyl imidate (5) (720 mg, 1.3 mM) and CaSO4 (400 mg) were added. After stirring at room temperature for 30 min. under nitrogen, TMSOTf (27 mg, 0.12 mM) was added, and the mixture was stirred at room temperature for 14 hours. After adding few drops of triethylamine, the mixture was filtered through celite, washed with dioxane, evaporated, and purified on a silica gel column (eluted with dichloromethane-methanol, 10:1). Each fraction was checked by LC-MS, unreacted acceptor (200 mg) was recovered and combined disaccharide mixture was dissolved into dry methanol (5 mL), and treated with MeONa/MeOH (1 M), stirred for 5 min., neutralized with Dowex 50 (H+) resin, filtered, evaporated, and the residue was purified on Iatrabeads with CH2Cl2/MeOH (3:1), to give the disaccharide library D as a white solid in 30% yield based on conversion of compound 1. Spectral data for library D: 1H NMR (D2O, 500 Hz): δ5.375 (d, J=2.3 Hz), 5.300 (d, J=2.2 Hz), 4.823 (d, J=2.2 Hz); ESIMS: m/z 618 [M+H]+.
Random glycosylation of per-O-benzyl galactosyl trichloroacetimidate (2) on LacNAc glycoside 6
To the mixture of LacNAc β-D-glycoside 6 (600 mg, 0.65 mM) and per-O-benzyl galactosyl trichloroacetimidate (2) (1300 mg, 1.3 mM) in dry DMF (5 mL) was added CaSO4 (500 mg). After stirring at room temperature under argon for 30 min., BF3•Et2O (29 mg, 0.13 mM) was added, and the mixture was stirred at room temperature for another 14 hours. Few drops of triethylamine were added to quench the reaction, and the reaction mixture was filtered through celite to remove the solid, washed with DMF, evaporated, and purified on a silica gel column elected with dichloromethane and methanol (10:1). Each fraction was checked by LC-MS, the unreacted acceptor, the donor hydrolysed products, and the trisaccharides were combined and hydrogenated in methanol in the presence of Pd/C for 48 hours at room temperature. After work up, the residue was purified on C-18 column with water and methanol to get the trisaccharide library E in 30% yield. Spectral data for library E: 1H NMR (D2O, 500 Hz): δ5.335 (d, J=2.3 Hz), 5.248 (d, J=2.7 Hz), 5.008 (d, J<2.0 Hz), 4.935 (d, J=2.4 Hz), 4.861 (d, J<2 Hz), 4.859 (d, J=2.5 Hz); ESIMS: m/z 780 [M+H]+.
As shown in Scheme 1 , 8-p-methoxyphenoxyoctyl-2-deoxy-2- acetamido-β-D-glucopyranoside was selected as an unprotected acceptor for the random glycosylation reaction.10 Its hydrophobic and UV active aglycon simplifies the purification of final products by C-18 column or HPLC. Schmidt’s glycosyl trichloroacetimidates (2-5) were used as donors for random gycosylation reaction.
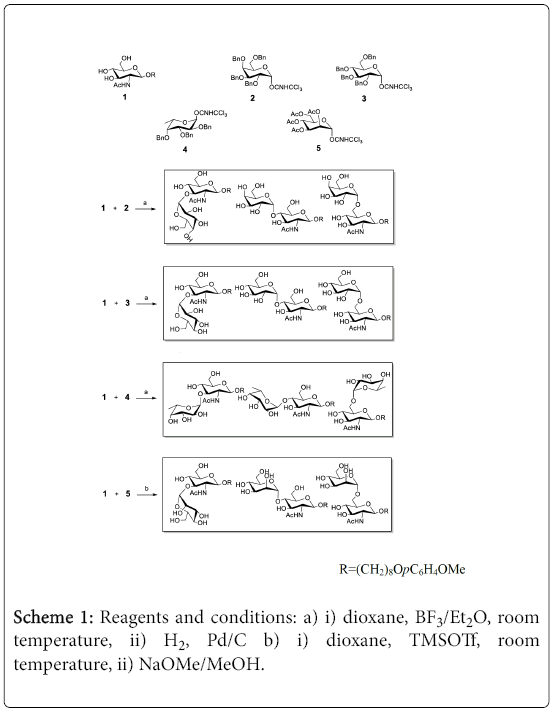
Scheme 1: Reagents and conditions: a) i) dioxane, BF3/Et2O, room temperature, ii) H2, Pd/C b) i) dioxane, TMSOTf, room temperature, ii) NaOMe/MeOH.
Accordingly, the Gal-GlcNAc disaccharide library was obtained from the coupling reaction of glycoside 1 and 2,3,4,6-tera-O-benzyl-α- D-galactopyanosyl trichloroacetimidate (2) (2 equiv.). The reaction was performed in dioxane at room temperature (3 hours) in the presence of 0.1 equivalent of BF3•Et2O. After addition of few drops of Et3N, the reaction mixture was carefully purified on a silica gel column, and each fraction was monitored by LC-MS.
From the reaction mixture, the desired disaccharides, unreacted acceptor, hydrolyzed derivatives of the donor, and trace amounts of trisaccharides were isolated. After hydrogenation of the disaccharide fraction, three disaccharides were obtained as a mixture in 25% yield (based on the conversion of compound 1). The proton 1D and 2D NMR spectra of the mixture indicated there are three α-linked disaccharides with 1:1:1 ratio (Figure 2).
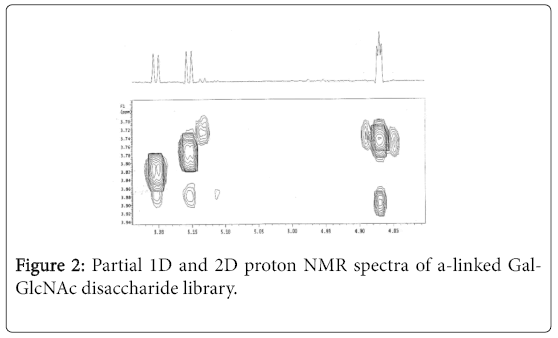
Figure 2: Partial 1D and 2D proton NMR spectra of a-linked Gal- GlcNAc disaccharide library.
The unprotected acceptor 1 contains one primary and two secondary hydroxyl groups, which may have different tendency towards glycosylation. However, under the given random glycosylation conditions, three possible α-linked disaccharides were formed in an acceptable ratio.
In order to improve the glycosylation yield, we attempted several different reaction conditions (such as changing the solvents and reaction temperature), but none of them improved the glycosylation yield any further.
Similarly, by using above glycosylation conditions and sequence of steps, starting from 2,3,4,6-tetra-benzyl-α-Dglucosyltrichloroacetimidate (3), 2,3,4-tri-O-benzyl-α-Lfucosyltrichrchloroacetimidate (4) and acceptor 1, we obtained the corresponding α-linked Fuc-GlcNAc and Glc- GlcNAc disaccharide libraries. The partial proton NMR spectrum of Glc-GlcNAc disaccharide library is shown in Figure 3, and the partial proton NMR spectrum of Fuc- GlcNAc disaccharide library is shown in Figure 4.
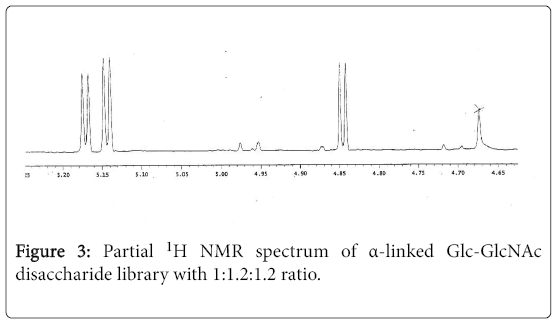
Figure 3: Partial 1H NMR spectrum of α-linked Glc-GlcNAc disaccharide library with 1:1.2:1.2 ratio.
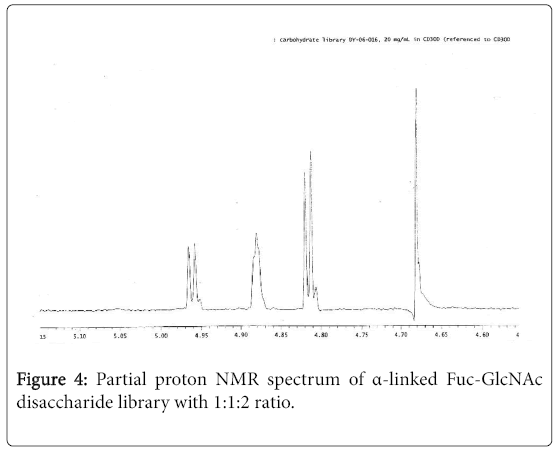
Figure 4: Partial proton NMR spectrum of α-linked Fuc-GlcNAc disaccharide library with 1:1:2 ratio.
Surprisingly, under the same reaction condition, synthesis of α- linked Man-GlcNAc disaccharide library through the coupling reaction of 2,3,4,6-tetra-O-acetyl α-D-mannosyltrichloroacetimidate (5) with compound 1 couldn’t be achieved, and BF3•Et2O was found to be a not suitable coupling agent for this reaction, and TMSOTf accomplished this task. After coupling reaction, purification and deacetylation, the final disaccharide library was obtained in 30% yield. The partial proton NMR spectrum of this library indicated all possible three α-linked Man-GlcNAc disaccharides were formed in 0.21:0.27:0.47 ratio (Figure 5).
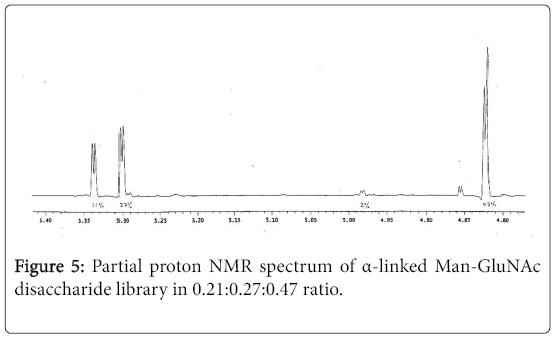
Figure 5: Partial proton NMR spectrum of α-linked Man-GluNAc disaccharide library in 0.21:0.27:0.47 ratio.
As an extension of the above random glycosylation strategy, random galactosylation on a LacNAc disaccharide acceptor was performed to generate an α-linked trisaccharide library (Scheme 2). Thus, the coupling of galactosylation donor 2 with the unprotected LacNAc 610 in DMF at room temperature in presence of BF3•Et2O was successfully achieved. After purification and hydrogenation, the trisaccharide library was obtained in 30% yield (based on the conversion of the unprotected donor). The proton 1D NMR spectrum indicated there are six α-linked trisaccharides were formed in 5:10:24:16:22:22 ratio (Figure 6).
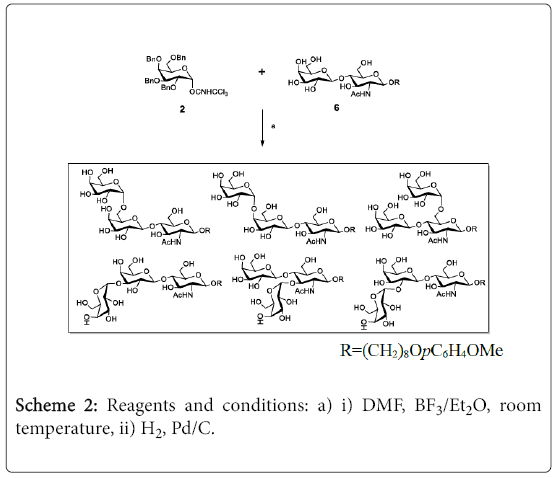
Scheme 2: Reagents and conditions: a) i) DMF, BF3/Et2O, room temperature, ii) H2, Pd/C.
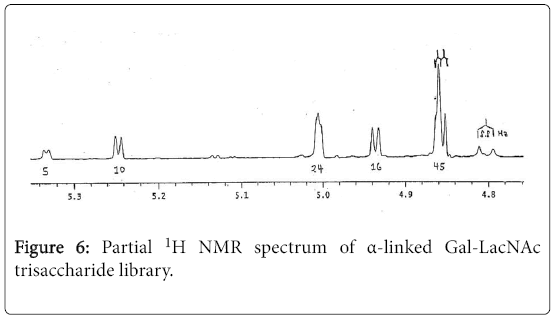
Figure 6: Partial 1H NMR spectrum of α-linked Gal-LacNAc trisaccharide library.
In conclusion, a concise one step method is developed for the synthesis of α-linked Gal-GlcNAc, Glu-GlcNAc, Fuc-GlcNAc and Man-GlcNAc disaccharide libraries in reasonable ratios. The method is also extended for galactosylation on unprotected LacNAc disaccharide to obtain the corresponding α-linked trisaccharide library. These libraries were confirmed by proton NMR data.