Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2009) Volume 2, Issue 9
Recently, genotypic testing (the identification of viral mutations and their associated drug resistance) has be- come a popular procedure to identify drug resistanc e before advising alternative therapy regimens.
Since major drug resistance factors are associated with the frequency of viral mutations, many researchers have explored HIV’s mutation frequency at specified nucl e- otide sequence positions in response to different t ypes of drug therapy. However, only a handful of papers dis- cuss major genetic signatures that lead to positive pa- tients’ responses to a specific type of drug therap y.
Using existing clinical drug resistance libraries, we were able to determine the most common mutations in the HIV protease (PR) enzymes associated with the s uc- cess and failure of Protease Inhibitor (PI) HIV/AID S drug regimens. A total of 2,079 patient records sel ected from the Stanford HIV drug resistance database has been considered in identifying genetic sequences associa ted with positive responses to PR-inhibitors drug regim ens. We show that patients who responded positively to P ro- tease Inhibitor therapy have consistently maintaine d specific nucleic acids bases at specific positions of their HIV nucleotide sequences. When virus sequences ob- tained from groups of patients who did not respond well to PI therapy were compared against virus sequences at the same positions from patients who did respond we ll, we noticed that the two patient groups differ at th ese positions.
Keywords: Drug resistance; Viral sequences; c2 test; CD4 count; RNA level; Viral envelop; Viral load assay
HIV: Human Immunodeficiency Virus; CD4: Cluster of Differentiation 4; RNA: Ribonucleic Acid
Major factors that determine the success of HIV therapy include the measurement and monitoring of CD4 counts, RNA (ribonucleic) levels, and the determination of the type of viral sequences and drug resistance mutations. CD4 is a primary receptor used by HIV-1 to gain entry into host T cells. The HIV-1 virus attaches to CD4 with a particular protein in its viral envelope known as gp120. The binding to CD4 creates a shift in the conformation of the viral gp120 protein allowing HIV-1 to bind to two other cell surface receptors on the host cell. Following another change in shape of a different viral protein (gp41), HIV inserts a fusion peptide into the host T cell that allows the outer membrane of the virus to fuse with the T cells membrane. HIV infection leads to a progressive reduction in the number of T cells possessing CD4 receptors and, therefore, the CD4 count is used as an indicator to help physicians and clinicians decide when to begin treatment in HIV-infected patients. Treatments often start when the CD4 count reaches a low point of around 200 cells per microliter. CD4 tests are also used to judge treatment efficacy. A novel method for counting CD4 in resource-poor settings has been described in (Rodriguez et al., 2005).
Another important indicator used to monitor HIV treatments is the viral load. Viral load assays include the measurement of HIV ribonucleic polymerase chain reaction (PCR), Roche Amplicor HIV-1 Monitor (Erali and Hillyard, 2005), the branched chain DNA using VERSANT HIV-1 RNA 1.0 Assay (kPCR) (Troppan et al., 2009), and nucleic acid sequence-based amplification NucliSens HIV-1 QT test (Ginocchio et al., 2003). Datasets considered in this research include RNA levels measured using the PCR qualitative approach. The main goal of HIV drugs is to reduce viral load as much as possible for as long as possible. Some viral load tests measure down to 400 or 500 copies of HIV per unit of blood; others go as low as 200 or even 50 copies. High levels--from 30,000 (in women) to 60,000 (in men) and above are linked to faster disease progression. Levels below 50 offer the best RNA reading.
Several sequence detection methods have been used to obtain viral sequences and detect drug resistance mutations. Major methods to obtain viral sequences include the 454 Life Sciences GS20 sequencing platform (Binladen et al., 2007) (which allows massively parallel picoliter-scale amplification), pyrosequencing of individual DNA molecules (Ronaghi et al., 1996; Nyrén, 2007), bidirectional sequencing (Mitsuya et al., 2008) and array sequencing (Miyada et al., 2008) (which has been used for sequence detection). Since different sequencing methods have different error tolerance, we selected patients’ RNA sequences obtained using the pyrosequencing sequencing procedure (Wang et al., 2007).
During the previous two decades, HIV data mining has taken different forms including modeling the rate of change of CD4 counts with respect to the change in RNA level and aligning a nucleotide or amino acid sequences for phylogenetic analysis tracing the epidemiology of HIV. However, it has become evident that the most efficient way to provide effective treatment analysis is by investigating viral drug resistance. Different forms of viral mutations take place due to the type and concentration of considered drugs, pre-treatment patients’ conditions (such as initial CD4 count and interaction with other medications), patients’ adherence, and demographic conditions. Such analysis can be used by clinicians to monitor and adjust patients’ health progress.
Four types of drug regimens are available to treat human immunodeficiency virus type 1 (HIV-1) infection (FDAWebsite, 2008): 1. The nucleoside reverse transcriptase inhibitors (NRTI) attempt to prevent the RT enzyme from changing the genetic code (RNA) into DNA by binding the RT enzyme to NRTI building blocks instead of the naturally occurring DNA blocks. 2. The non-nucleoside reverse transcriptase inhibitors (NNRTI) attempt to inhibit the RT enzyme by reducing its conformational flexibility. 3. Protease inhibitors (PI) prevent the protease enzyme from assembling functioning virus from a raw material HIV virus. 4. The fusion inhibitors prevent HIV from attaching to a cell (only one fusion inhibitor, Enfuvirtide, has been approved).
Each of the above treatment methods can be rendered ineffective by one or more types of drug resistance (Johnson et al., 2008). HIV drug resistance occurs when HIV changes or mutates such that an antiretroviral drug loses its effectiveness to stop the spread of the virus. HIV viral mutations result from errors in duplicating the virus' genetic code and hinders the development of therapeutic drugs that can eliminate or restrain the process of viral duplication (Sandstrom et al., 2008). In addition to mutations resulting from errors in replication of genetic information, other types of HIV drug resistance mutations may occur in response to a specific therapeutic drug. For example, the single mutation at position 184 involving a transition from methionine to valine (M184V) mutation is particularly associated with the 3TC (Lamivudine) treatment (Diallo et al., 2003). Attempts to study HIV sequence mutations using sequence alignments techniques also showed adequate success (Phillips et al., 2008). An up-to-date and comprehensive survey for major mutations associated with NNRTI, NRTI, and PI drugs can be found in (Johnson et al., 2008; Shafer and Schapiro, 2008). However, only a few research papers discuss major genetic signatures that lead to positive patients’ response to a specific type of drug therapy.
Due to the drastic effects of drug resistance on the effectiveness of HIV therapy, it has become important for physicians to use genotypic resistance data (protease and RT mutations) in making decisions regarding the best therapy that avoids drug resistance. The most common use of drug resistance datasets is to use the HIV virus' genotype and phenotype datasets to interpret HIV mutations in response to anti-retroviral therapy (Rhee, 2007; Wrin et al., 2000). This aids in the determination of appropriate regimens using drug resistance databases such as those developed in (Rhee et al., 2003; Vondrasek and Wlodawer, 2002; Macke et al., 2007).
Many online HIV databases such as (Los Alamos National Laboratory, Rhee et al., 2003; Graziano et. al., 2008; NIAID Division of AIDS, University of California; San Francisco, HIV InSite) are available to obtain mutation frequencies and positions due to drug resistance. Using these databases, researchers and clinicians can obtain patients’ datasets including sequence database, resistance database, immunology database, and vaccine trial database. Analyses tools are provided in most online databases to obtain relevant correlations such as genotype-treatment correlations, genotype-clinical correlations, and genotype-phenotype correlations. Main objectives for using online databases would be to obtain underlying data and references that show which mutations cause resistance and to which drugs (Rhee et al., 2003), obtain statistical analyses such as correlations between CD4 and RNA levels for considered treatments (Graziano et al., 2008), obtaining rate of change of CD4 and RNA levels based on base CD4 count at the start of a considered patient or a considered therapy, and obtain rate of change CD4 and RNA levels based on preceding CD4 counts for a considered patient or a considered therapy (Graziano et al., 2008).
One of the most comprehensive HIV drug resistance libraries is the Stanford HIV database developed by (Rhee et al., 2003). This library contains genetic data on HIV isolated from more than 10,000 individuals including about 2,000 individuals from Northern California described in recently published studies. Each patient record consists of a patient's genotypes, treatments, plasma HIV-1 RNA levels, and CD4 counts at various clinical trials. Patients are classified, among anti-retroviral treatment-naive HIV-1 infections, according to the prevalence of mutations associated with anti-retroviral drug resistance in protease (PR) and reverse transcriptase (RT) regions.
In this work, we use 2,079 patient records available in the Stanford drug resistance database to generate lists of genetic sequences for patients responding well to drug regimens. We compare those sequences to HIV sequences obtained from patients with poor response to HIV drug regimens. Statistical analysis is provided for RNA locations that indicate significant differences between positive and negative drug responses. We aim to satisfy the following objectives:
1. Determine major mutations associated with the success and failure of common HIV drug therapies.
2. Develop a software package that can be used to predict (and explain) the probability of success or failure of a given drug using historical patients' records in association with available genetic datasets obtained from the two libraries.
3. Provide genetic sequence positions where a patient's positive response to PI drug regimens differs from patients who did not respond well to PI regimens.
To prepare our datasets, we selected all patients with consistent improvements of CD4 values and decline of the RNA levels. Out of 2,079 records stored in the Stanford PR database, we found 189 patient IDs with monotonically non-decreasing CD4 counts and increasing RNA levels throughout their clinical trial history. We then selected ten sets each with 189 randomly chosen patients that did not satisfy the monotonically non-decreasing criteria. For each patient in the eleven chosen datasets, we selected viral RNA mutations sequences associated with the selected patients’ treatments. At the end of this process, we obtained one dataset containing 189 sequences for patients who responded well to their drug regimens, and ten datasets each containing 189 sequences representing patients who did not respond well to their drug regimens. Figure 1 depicts a selected patient's nucleotide RNA sequence file with the corresponding CD4 counts and RNA levels. We used Eq. (1) to calculate the probability distribution of each base (A, C, T/U, G) in each of the 297 nucleic acids bases’ positions among the 189 sequences:
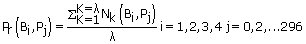 (1)
(1)
In Eq. (1), λ represents the total number of considered sequences (λ = 189), k represents a considered sequence number (k=1…189), and j represents a base position for each of the 297 bases (in each of the 189 sequences). represents the probability distribution of base (A, C, T/U, G) at position . The operator Nk(Bi, Pj) produces a value of one if base i exists at position j of sequence k, otherwise, it produces a zero. At the end of the probability distribution calculation process, we obtained 11 matrices, each with 297 rows (representing the 297 base positions) and 4 columns (representing the probability distribution of each of the four bases, A, C, T/U, and G as calculated from Eq. (1)). Table 1 [Table 1 about here] depicts part of the 297 4 probability distribution matrix resulted from Eq. (1)’s implementation using 189 patients’ nucleic acids sequences (this matrix represents data for patients who satisfy the monotonically increasing CD4 counts criteria). The total probability distribution at each row of Table 1 might not equal to one due to the unavailability of sequenced datasets at different positions (Stanford database library uses the * symbol to indicate unrecognized nucleic acid bases).

Figure 1: Nucleotide sequence, CD4 count, and RNA level for patient 23438 for three treatment trials (weeks 0, 4, and 7).
| A | C | T | G | |
|---|---|---|---|---|
| P0 | 0 | 0.687 | 0 | 0 |
| P2 | 0 | 0.687 | 0 | 0 |
| P3 | 0 | 0 | 0.687 | 0 |
| P4 | 0 | 0.693 | 0 | 0 |
| P5 | 0.693 | 0 | 0 | 0 |
| P6 | 0.216 | 0 | 0 | 0.449 |
| . | . | . | . | . |
| . | . | . | . | . |
| P296 | . | 0.021 | 0.962 | 0 |
Table 1: Part of the results obtained from Eq. (1) to calculate the probability distribution values of A, C, T/U, G bases at different nucleotide acid positions using patients’ sequences that satisfy the monotonically CD4 increase and RNA decrease criteria.
Table 1 shows that all patients satisfying the 100% CD4 increase and RNA decline criteria do not have Adenine at their first nucleic acid sequence positions. Looking at P0, we can deduce that there are 130 C bases in the first position of those 189 sequences (we truncated the probability distribution to three decimals). The probability distribution of the T base at position 296 is 0.962963, which indicates that, given that we have 189 sequences, there are 182 T bases at position 296 of the 189 RNA sequences.
Having obtained the eleven probability distribution matrices, we implemented the χ2 test to verify if the ten randomly selected nucleic bases distributions, obtained from the probability distribution matrices for patients with minimum response to the PI therapy, differ from the distribution of the nucleic sequences for patients who showed good response to PI therapy. Significant dependency between the two patients’ groups will reduce our datasets and, hence, minimize our ability to use them for further analysis. To determine how much each of the four nucleic acids (A, C, T/U, and G) contributed in the rejection of the null hypothesis, we calculate the χ2 value (with a p-value of 7.82 and α-value of 0.05 for three degrees of freedom for the four types of nucleic acids bases) for the four nucleic acids. Figure 2 and Table 2 (that summarizes Figure 2 data) show that in seven of the ten independent sets, Adenine contributed in the indecency (in distribution) of the two datasets at positions 187 and 267. Thymine has contributed significantly in seven of the ten randomly selected datasets at position 59. The figure and table also show base-positions that contributed significantly to the dependency of the ten randomly selected datasets.

Figure 2: The results of χ2 test. Ten randomly selected patients’ datasets (of CD4, VL, and nucleotide sequences) passed the null hypothesis test with χ2 of 7.82 and p-value of 0.05 for three degrees of freedom (for the four types of nucleic acids bases).
| Frequency of significant occurrences (in ten random sets) | Adenine nucleotide positions | Cytosine nucleotide positions | Thymine nucleotide positions | Guanine nucleotide positions |
| 7 | 187, 267 | 59 | ||
| 6 | 108 | 213, 216 | 104 | |
| 5 | 5, 183, 186, 211 | 219 | 183, 278 | 46, 275 |
Table 2: Adenine contributed significantly at positions 187 and 267 to the independence of seven of the ten considered datasets.
Having determined the independency of the distribution of the ten selected datasets from datasets associated with patients who showed positive response to the PI regimens, we proceeded to investigate the type and position of nucleic acid bases that differ most between the two types of datasets at each nucleic acid position. We used the calculated 11 [297 4] probability distribution matrices (one for each of the eleven datasets) to calculate the variation between the monotonically increasing CD4 count probability distribution matrix and each of the ten randomly selected patients’ probability distribution matrices that do not satisfy the monotonically increasing CD4 count criteria. To do this, we calculated Euclidean distances between each probability distribution value in the monotonically increasing CD4 count probability distribution matrix and each of the other ten randomly selected non-monotonically increasing CD4 count probability distribution matrices. We thus obtained ten Euclidean distance matrices. Although in what follows we show detailed Euclidean distances calculations for the A bases, all assumptions and equations are applicable to the other three bases (C, T, and G).
Assuming that αAi represents the probability distribution of base A at position i in the consistently increasing CD4 counts file, and βAi represents the probability distribution of base A at position i in the non-monotonically increasing CD4 counts datasets, we can calculate the square of Euclidean distance for base A at position i (EAi) using the formula:
EAi = (αAi - βAi)2 (2)
We considered EAi values that display a 95% match between αAi and βAi to be significant values. Therefore, we assigned any EAi value greater than 0.025 to 1 (to indicate a significant difference between αAi and βAi ) and those values less than 0.025 are assigned to 0 (to indicate non- significant differences). Thus, if SAi represents the significance of a considered Euclidean distance EAi then:
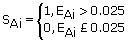
Having evaluated SAi values for all EAi we obtained ten column values (each with 297 entries) representing significant (represented by 1) or non-significant (represented by 0) distances between αAi values and each of the ten sets of the βAi values. The same procedure was applied to the significant matrices for each of the other three types of nucleic acids bases C, T/U, and G.
Figures 3, 4, 5 and 6 represent average SAi, SCi, STi, and SGi values at each of the 297 RNA positions. Nucleic acid positions shown in each figure represent each of the SAi, SCi, STi, and SGi values calculated using Eq.(2). Values close to 1 indicate a 100% difference between the number of considered type of nucleic acid calculated for patients who responded positively to the PI regimens in comparison to those who did not respond well to the same PI regimens (at a given position). For example, Figure 4 shows that while Adenine exists in position 6 in the all positively responding patients, it does not exist at position number 6 in any of the negatively responded patients. Figure 7 summarizes the results depicted in Figures 4, 5, and 6. We chose to display base positions where 77-100% significant differences occur between positively responding patients and other patients. It is clear from the figure that while position 6 displays a 100% (represented here by A_1) difference as far as the existence of Adenine in both patient groups, a significant 80% differences (represented by A_8) are calculated between the two groups, for Adenine, at positions 9, 27, 169, 224, and 276. Furthermore, 88% of the C base has changed between the two patient groups at positions 3 and 8, and 77% of bases showed differences from the C base at positions 0, 104, and 211. Similarly, unlike patients who responded well to PI regimens, 100% of patients who did not respond well to the PI regimens did not have a T/U base at position 255. Additionally, 88% of the sequences did not have T/U at positions 149, 272 and 287. Finally, Figure 7 shows that 100% of patients who did not respond well to the PI regimens did not have G in positions 206 and 273 (unlike patients who responded well to the PI regimens where G exists at these positions).

Figure 3: Average Euclidean distances randomly selected from ten datasets, each containing 189 patients. Euclidean distances are calculated between Adenine positions in patients who showed positive reaction to PR-inhibitor drug therapy, and the corresponding Adenine positions for patients who did not react positively to PR treatments. Positions displayed at level of 1 indicate significant differences from positive therapeutic reaction (hence, these positions are conserved in strains that do not develop PR-inhibitor resistance).

Figure 4: Average SCi values at each of the 297 RNA positions (C=Cytosine).

Figure 5: Average STi values at each of the 297 RNA positions (T=Thymine/Uracil).

Figure 6: Average SGi values at each of the 297 RNA positions (G=Guanine).

Figure 7: Adenine, Cytosine, Thymine and Guanine nucleotide positions where patients with positive response to PI drug regimens have 77-100% Euclidean distance from patients with negative PI response. In the figure, A_1:6, for example, indicates that 100% (represent as A_1) Adenine has been detected at position 6 in both monotonically increasing CD4 dataset while it has not been detected at this position in all the ten randomly selected non-monotonically increasing datasets. Also, A_0.8 indicates that Adenine depicts an 80% difference between the two sets at positions 9, 27, 169, 224, and 276.
The results discussed in this paper show that certain features in the HIV genome may be exploited to identify HIV strains that are highly susceptible to certain drug regiments and have a reduced chance of developing drug resistance. As shown in this paper, an Adenine nucleoside in position 6 signifies a virus that is less likely to become resistant to PR inhibitors. Furthermore, an Adenine in positions 9, 27, 169, 224, 276 or Thymine/Uracil at positions 255, 149, 272, 287 and Cytosine at positions 0, 3, 8, 104 and 211 may also signify similar behavior. The method developed here can be used to detect the susceptibility of an HIV strain to PR inhibitors and other types of anti-viral drugs in patients. Further studies conducted on larger sequence databases may also provide validation and more conserved motifs/signatures that can be very useful for identifying the susceptibility of HIV strains in patients. Future work in this area will focus on the identification of nucleoside sequences (whole motifs) instead of single nucleosides that may be conserved among strains that are less likely to develop drug-resistance.
The first three authors would like to thank the Grow Iowa Values Fund (GIVF) for funding this project. The authors would like to thank Dr. Robert Shafer from Stanford University Medical Center for his valuable input and for providing access to the hivdb.stanford.edu’s patients’ datasets. The authors appreciate this paper’s reviewers for their valuable comments.