Journal of Fertilization: In Vitro - IVF-Worldwide, Reproductive Medicine, Genetics & Stem Cell Biol
Open Access
ISSN: 2375-4508
ISSN: 2375-4508
Research Article - (2015) Volume 3, Issue 2
Embryonic stem (ES) cells have the ability to maintain pluripotency and self-renewal during in vitro maintenance, which is a key to their clinical applications. ES cell quality has been widely evaluated through the determination of their specific genetic and epigenetic profiles. The hypothesis of this study is that genetic stability in repetitive sequences located near key genes involved in pluripotency, self-renewal, differentiation, chromatin assembly, and imprinting could be a signal for adaptation of the ES cell in vitro. Instability in specific repetitive sequences is present and increases during ES cell passages. ES cells displayed significant mean frequencies of instability in twelve markers out of 64 related to pluripotency (OCT4, D1S551), early differentiation (G60405, D18S63, and D1S468), chromatin assembly (D22S447, D6S2252, D10S529, and HISTB2), and imprinting (GRB10-promoter, D2S144, and IGF2- promoter). Interestingly, instability was different between H1 and H7 ES cell lines. In summary, these results suggest that instability in tandem repeat sequences located near early embryonic developmental genes is associated with failure of ES cell pluripotency and self-renewal maintenance over consecutive culture passages. These results suggest that instability determination is a potential indicator of gene deregulation and epigenetic modification that involves chromatin modification and imprint establishment during ES cell culture. Finally, instability in specific genes could be a signal that contributes to the adaptation of ES cells to in vitro culture or could be the switch that initiates early cell specialization in vitro.
<Keywords: Genomic instability; Embryonic stem cells;Genome repetitive sequences; Self-renewal biomarkers
Since the first human embryonic stem (ES) cells were isolated two decades ago, this field of research has generated countless advances and knowledge about early embryonic development and cell fate differentiation [ 1-4]. Studies of ES cell pluripotency and self-renewal as the source of all cell types from the three embryonic germinal layers led to significant discoveries and clinical applications. However, continued maintenance in vitro leads to cellular, genetic, and epigenetic changes in the ES cells, which creates many questions about their real therapeutic potential. The accepted culture conditions used for ES cell maintenance around the world are limited due to different protocols between laboratories. Because of the wide range of variability in the maintenance of homogeneous and undifferentiated ES cells over time during culture passages, the clinical importance of ES cell research is sometimes doubted [5,6].
Several studies have reported changes in ES cell gene expression profiles that occur during long term culture [3,7,8]. Also, the presence of chromosomal abnormalities in late passage cultures of ES cells has been reported [9-13]. Furthermore, the signals or initial steps that lead to gene expression and epigenetic changes remain unknown. A simple screening method to select the best ES cells would be of great use in the field. This study focuses on determining the role of instability in repetitive DNA sequences as a signal of ES cell adaptation or differentiation, and the identification of possible biomarkers useful for screening and determining the quality of ES cells to be used for regenerative therapies.
Instability in flanking regions of developmental genes could affect enhancer or repressor elements that regulate transcriptional patterns of ES cells during in vitro maintenance. In order to understand how genomic instability affects pluripotency of ES cells, self-renewal, and differentiation, we have used a key characterization method to evaluate the effect of the ES cell. As a first step, we have investigated the instability effects of repetitive sequences on ES cells over time, and also we have determined the mean frequency of instability in different markers located close proximity to sequences of important genes responsible for ES cell pluripotency, self-renewal, cell differentiation, chromatin assembly, and imprinting. We have analyzed H1 and H7 ES cell lines during early, middle, and late passages to compare the genomic instability across passages. By determining the mean frequencies of instability for each marker, we identified sensitive repetitive markers that showed significant instability in ES cell cultures over time. In addition, specific genes that were identified as related to the unstable marker were evaluated. This study has established that instability in these specific regions could modulate gene expression and epigenetic signals that determine ES cell adaptation or differentiation stages.
Embryonic stem cell maintenance
Frozen aliquots of human ES cells H1-WA01 passage 27 and H7- WA07 passage 26 were purchased from the National Stem Cell Bank Wisconsin International Stem Cell Bank. H1 and H7 ES cells were seeded onto a mouse embryo fibroblast-CF1 (MEF) feeder layer previously inactivated with mitomycin C. The culture medium consisted of Dulbecco’s Modified Eagle Medium (DMEM) knockout medium (Invitrogen, Carlsbad, CA) supplemented with 20% knockout serum replacement (Invitrogen, Carlsbad, CA), 1% antibiotic-antimycotic (Invitrogen, Carlsbad, CA), 100 mM L-glutamine (Invitrogen, Carlsbad, CA) plus β-mercaptoethanol, 2 ug/ml basic fibroblast growth factor (b-FGF) (Invitrogen, Carlsbad, CA), and 1% non-essential amino acids (Invitrogen Carlsbad, CA). ES cells were maintained in a humidified atmosphere at 37°C in 5% CO2. Medium was changed daily.
Mouse embryo fibroblast CF1 feeder layer
The mouse embryo fibroblast (MEF-CF1) feeder layer cells were purchased from American Type Culture Collection (ATCC, Rockville, MD). MEF feeder layer cells were cultured in a T-25 flask (Falcon, Becton Dickinson Labware, NJ). The culture medium consisted of DMEM high glucose medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA) and 1% antibiotic-antimycotic (Invitrogen, Carlsbad, CA). MEF cells were mitotically inactivated for 2 hours with 10 μg/ml mitomycin C (Sigma Aldrich, Saint Louis, MO), seeded at densities of 130,000 cells/ml in gelatin coated one-well dishes (Falcon, Becton Dickinson Labware, NJ) and cultured 24 to 48 hours before ES cells were seeded onto the feeder layer. These cells were maintained in a humidified atmosphere at 37°C in 5% CO2.
Embryonic stem cell passages
ES cell colonies with undifferentiated morphologies were mechanically dissected into small pieces under a stereomicroscope and seeded onto a fresh MEF feeder layer during 20 passages (5 months). Cells were passaged every 4-6 days (Supplementary Material Figure S1). Periodically, ES cells were tested for the presence of alkaline phosphatase activity, which is an indicator of the undifferentiated state. We used the alkaline phosphatase detection kit following the manufacturer’s recommended protocol (Millipore, Chemicon, Billeria, MA). Samples of ES cell colonies were dissected for isolation of DNA and RNA early in the culture time (passage 27-28) and during the middle of the culture time (passage 40-42) in both ES cell lines.
Immunohistochemical analysis
ES cell colonies were fixed with 4% paraformaldehyde (Sigma Aldrich, Saint Louis, MO) for 15 minutes at room temperature, washed in PBS, and immunostained. The primary antibodies used were rabbit anti-OCT4 polyclonal antibody, mouse anti-SOX2 monoclonal antibody, and mouse anti-SSEA-1 alexa fluor 488 (Chemicon/Millipore, Billerica, MA). Secondary antibodies included goat-anti-rabbit IgG rhodamine, and CY5-conjugated antibody (Chemicon/Millipore, Billerica, MA). Each antibody was diluted 1:200 in PBS, 0.1% Triton X-100, and 3% BSA. Nuclei were visualized with 4’-6-diamidino-2- phenylindole (DAPI) staining (Vysis Abbott Laboratories, Abbott Park, IL). Staining without primary antibody served as a negative control. Images were captured using a fluorescence microscope Axiovert 135 (Carl Zeiss International) with FITC and rhodamine filter set. Fluorescence intensities were measured with image software developed at the National Institute of Health (Bethesda, MD) downloaded from http://rsb.info.nih.gov/ij/index.html.
DNA isolation
ES cells colonies before isolation of DNA were passage into matrigel plates for eliminate MEFs contamination. DNA was prepared from each sample of ES cells in early passage (27-28) and middle passage (40-42). DNA from late passage (78-82) was provided by the Michigan Center for human ES Cell Research (Ann Arbor, MI) who followed the same protocol for maintenance and passages of cells like our research. DNA was isolated with the Purelink genomic DNA mini kit (Invitrogen Carlsbad, CA) following the manufacturer’s protocol. All DNA samples were quantified using a NanoDrop™ ND1000 spectrophotometer (Thermo Scientific, Wilmington, DE).
Single tandem repeat markers selection and standardization
Single tandem repeats (STRs) are located in or near promoter regions of specific genes responsible for embryonic stem cell pluripotency and self-renewal. We identified DNA sequences that were approximately 1000 bp upstream or downstream of the promoter using UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/) gene sorter and uni-STSNCBI database (http://www.ncbi.nlm.nih.gov/genome/sts/sts.cgi). A total of 64 STR were selected and classified according to ES genetic network regulation database available at (http://www.wi.mit.edu/ young/hESregulation/). Eleven markers were related to pluripotency genes, 33 were related to differentiation genes, 12 were related to chromatin modification genes, and 8 were related to imprinting genes (Table 1). Each STR was optimized to obtain amplified products with robust signal intensity and balanced peak heights from ES cell samples in early passage (27-28), middle passage (40-42), and late passage (78- 82).
| Pluripotency | Differentiation | Chromatin Structure | Imprinting | |
|---|---|---|---|---|
| OCT4 | D16S3034 | D4S1542 | D7S488 | GRB10PROM |
| D1S1656 | D12S1719 | DXS981 | D6S1001 | D20S821 |
| D1S551 | D4S2623 | D14S588 | HISTH4A | IGF2R |
| D12S1682 | D2S134 | D3S2459 | HISTHB2 | DIRAS3PROM |
| D1S2630 | D11S1331 | D17S2180 | D10S529 | PEG10PROM |
| D6S2384 | D4S1625 | EGFR | D22S447 | SNURF10PROM |
| D6S416 | D1S430 | D16S3091 | D8S11268 | IGF2PROM |
| D2S2327 | D2S290 | D1S468 | D22S941 | IGF |
| kLF4-1 | D3S1583 | TNFa3 | D7S638 | |
| NANOG | DXS458 | D15S983 | D6S2252 | |
| D9S1840 | D21S1909 | DXS1208 | D2S144 | |
| D6S1698 | D5S426 | DNMT3 | ||
| D10S1653 | D3S1541 | |||
| D11S909 | G60405 | |||
| D5S2021 | D3S1611 | |||
| D18S63 | D11S2179 | |||
Table 1: List of single tandem repeat marker Eleven markers were related to pluripotency genes, thirty-three were related to differentiation genes, twelve were related to chromatin structure genes, and eight were related to imprinting genes
Samples were analyzed with differing amounts of genomic DNA: large DNA concentration (DNA concentration of 0.1 to 1 ng/μl) and single cell DNA concentration (single genome equivalent between DNA concentrations 12.5 to 50 pg/μl). The average for amplifiable DNA (λ) was calculated by Poisson distribution: λ= - ln (number of replicates with non-amplification / total number of replicates) [80]. A λ < 2 means that single genome equivalent of DNA was present in the amplification.
Each locus was standardized in separate PCR reactions to optimize and ensure specificity and sensitivity of the system. Labeled primers with either 6-FAM or HEX dye were used to allow automatic detection. Primers were tested at concentrations of 0.8-1.5 μM in standard PCR conditions and reagents.
Genomic instability determination by single cell PCR
Single cell PCR was performed on 64 STRs (Table 1). Less than a single diploid genome-equivalent of DNA (25-50 pg/μl), was used to perform single cell PCR analysis in 48 replicates for each marker. These concentrations of DNA ensure sensitivity of the PCR to detect wild type and mutated alleles at their appropriate frequency [14]. Total reaction volume of 10 μl containing 1X of buffer D (800 mM Tris HCL, 200 mM (NH4)2SO4, 0.2% w/v Tween 20) (US DNA, Fort Worth, TX), 2.5 mM of MgCl2 (US DNA, Fort Worth, TX), 1.25 U of Hot-MultiTaq DNA polymerase 5 U (US DNA, Fort Worth, TX), 4% of DMSO (Sigma Aldrich, Saint Louis, MO), 0.4 mg/ml of BSA (Thermo Scientific, Rockford, IL), 300 μM of dNTPs mix (Applied Biosystems, Foster City, CA), and 1X of Solution L 5X (enhancer solution for amplification of difficult templates) (US DNA, Fort Worth, TX). The primer volumes for each primer are shown on Supplementary Material Table S1.
PCR was performed on a PE 9600 thermocycler using a ramping protocol: 1 cycle of 95°C for 11 minutes; 1 cycle of 96°C for 1 minute; 10 cycles of [94°C for 30 seconds, ramp 68 seconds to 58°C (hold for 30 seconds), ramp 50 seconds to 70°C (hold for 60 seconds)]; 25 cycles of [90°C for 30 seconds, ramp 60 seconds to 58°C (hold for 30 seconds), ramp 50 seconds to 70°C (hold for 60 seconds)]; 1 cycle of 60°C for 30 minutes for final extension; and hold 4°C. Negative controls per run were included to check for contamination.
Amplified products were mixed with Hi-Di™ formamide and GeneScan™ 500 LIZ Size Standard (35-500 bp) (Applied Biosystems, Foster City, CA) and denatured for 3 min at 95°C to be separated and detected by fragment analysis on a Genetic Analyzer AB3130xl (Applied Biosystems, Foster City, CA). Data were analyzed with the software, GeneMapper version 4.0 (Applied Biosystems Foster City, CA). Quantification of the allele size in comparison with the internal lane size standard was scored in each single cell replicate. An average of 48 replicates per sample plus negative controls were amplified and scored for both ES cell lines.
STR makers are classified according to their repeat motif (number of nucleotides): mononucleotides (1 nucleotide motif), dinucleotide (2 nucleotide motif), trinucleotide (3 nucleotide motif), tetranucleotide (4 nucleotide motif), and pentanucleotide (5 nucleotide motif). Wild type alleles were determined for each microsatellite. Repeat motif shifts from the wild type allele size were considered a mutant allele. Mutant alleles for mononucleotides (e.g. GRB10-PROM, IGF2-PROM, and HISTBH2) were determined by a repeat shift greater than 3 repeats or less than 3 repeats. For dinucleotides (e.g. D18S63, D6S2252, and D10S529), mutants were determined by a repeat shift greater than 2 repeats or less than 3 repeats. For trinucleotides (e.g. D17S2180), tetranucleotides (e.g. OCT4, and D1S551) and pentanucleotides (e.g. DIRAS3-PROM), mutants were determined by a repeat shift greater than 1 repeat or less than 2 repeats (Supplementary Material Figure S2) [14-18].
Statistical analysis of genomic instability
Mutation frequencies (total number of wild type alleles related to the mutant alleles in each marker) were determined for each ES cell line and passage number by SP-PCR software version 2.0 (M.D. Anderson Cancer Center Houston, TX). Differences in mutation frequencies were calculated with a two tailed t-test using raw mutation frequencies using a package SAS/win 9.2 (SAS Institute, Cary, NC). Mutation frequencies of informative markers were considered statistically significant when a p-value was ≤0.05, and were considered marginally significant if the p value was ≤ 0.10
Embryonic stem cell culture maintenance
ES cells were continuously cultured for 20 passages to explore the potential role of genomic instability during ES cell maintenance in vitro under standard conditions with MEFs and growth factors (b-FGF). ES cells from both cell lines (H1 and H7) retained their growth and morphological characteristics: homogenous round and compact colonies, a prominent nucleus and high nucleus: cytoplasm ratio, positive alkaline phosphatase activity and positive expression of the specific pluripotency marker OCT4 and negative expression of the differential marker SSEA-1.
Embryonic stem cells displayed morphological changes across passages
ES cell cultures, in general, could contain fewer than 20% of colonies with heterogeneous morphology corresponding to differentiation. These heterogeneous colonies were removed with a pipette using a stereomicroscope before the subsequent passage [19-21]. H7 ES cells were subcultured more than 20 times continuously for more than 5 months. During that time, they exhibited round and compact colony morphologies. In contrast, H1 ES cells were cultured under the same conditions and time, yet they exhibited an increased number of irregular shapes of colonies with some differentiated cells at the periphery (Figure 1). To explore the ES cell morphological characteristics over passages, we compared differences in the shape of the colonies between H1 and H7 ES cells; we quantified the number of regularly and irregularly shaped colonies from passages 28-42 in H1 ES cells and 27-42 in H7 ES cells. We found that H1 ES cells showed a significant increase in the colonies that exhibited signs of cell differentiation across passages in comparison to the H7 ES cell line (p=0.04). H1 ES cells in passage 40 showed a higher percentage (37%) of irregular colonies than did those from passage 27 (14%) (p=0.047) (Figure 2). The H7 cell line did not show any significant difference across passages. Taken together, these results indicate that H1 ES cells failed to promote complete self-renewal across passages.
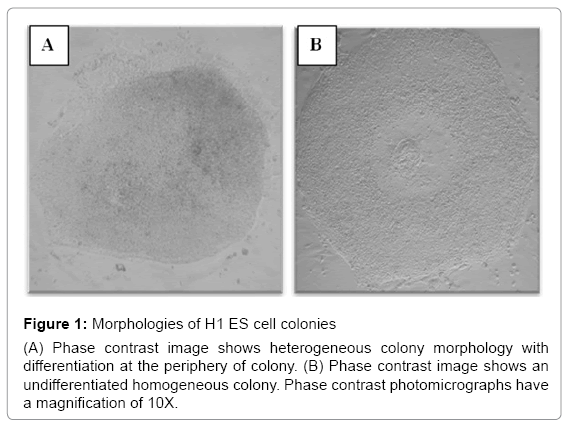
Figure 1: Morphologies of H1 ES cell colonies
(A) Phase contrast image shows heterogeneous colony morphology with differentiation at the periphery of colony. (B) Phase contrast image shows an undifferentiated homogeneous colony. Phase contrast photomicrographs have a magnification of 10X.
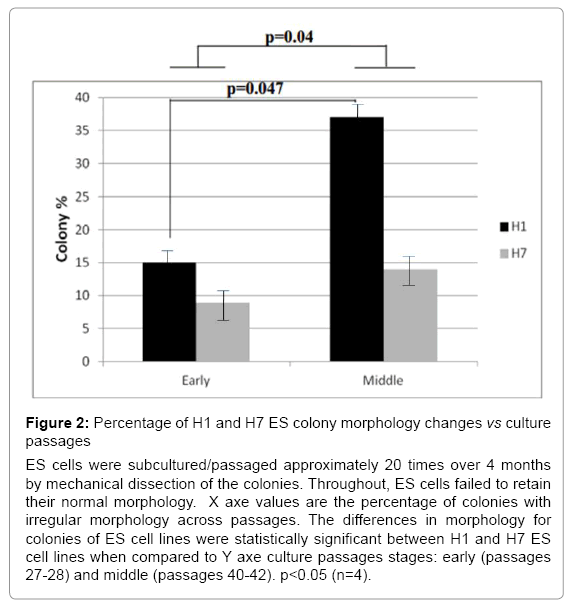
Figure 2: Percentage of H1 and H7 ES colony morphology changes vs culture passages
ES cells were subcultured/passaged approximately 20 times over 4 months by mechanical dissection of the colonies. Throughout, ES cells failed to retain their normal morphology. X axe values are the percentage of colonies with irregular morphology across passages. The differences in morphology for colonies of ES cell lines were statistically significant between H1 and H7 ES cell lines when compared to Y axe culture passages stages: early (passages 27-28) and middle (passages 40-42). p<0.05 (n=4).
Genomic instability in single tandem repeat markers was associated with embryonic stem cell culture adaptation
Because embryonic stem cells in culture maintain pluripotency and self-renewal via genetic rearrangements [3,9-13], we asked whether ES cell cultures are genetically stable in long term cultures. The efficiency of ES cells to maintain genomic stability was evaluated by analyzing single tandem repeat markers found close to specific genes involved in ES cell pluripotency and self-renewal (Table 1). Samples of DNA from H1 and H7 ES cells at three different times (early, middle, and late passages) were analyzed to determine genomic instability in specific markers. There was significant genomic instability in 21 out of 64 single tandem repeat markers evaluated. Both ES cell lines were unstable in these markers over cell passages. However, H1 ES cells became much more unstable than H7 ES cells. H1 ES cells showed significant instability differences between early to middle (p=0.002) and between early to late passages (p=0.025) but differences were not significant between middle to late passage. In contrast, H7 ES cells show a significant difference only between early to middle passage (p=0.057) (Figure 3). These results indicate genomic instability was present during long term ES cell cultures and suggest these could be a signal of cell adaptation.
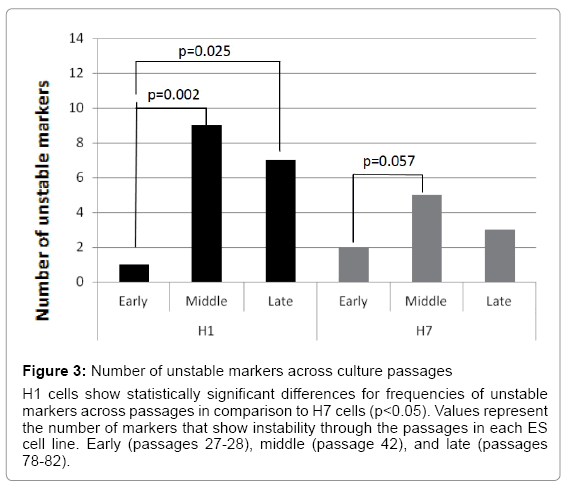
Figure 3: Number of unstable markers across culture passages
H1 cells show statistically significant differences for frequencies of unstable markers across passages in comparison to H7 cells (p<0.05). Values represent the number of markers that show instability through the passages in each ES cell line. Early (passages 27-28), middle (passage 42), and late (passages 78-82).
Genomic instability could be a signal of embryonic stem cell pluripotency and self-renewal loss during long term cell culture
Increasing evidence suggests that culture passages of ES cells lead to significant changes in gene expression [3,7,8,22]. Our results have shown that during long term culture and subsequent passages, ES cells accumulated instability in single tandem repeats. These markers are located near important genes involved in pluripotency and differentiation. H1 ES cells were unstable in three markers related to pluripotency genes (OCT4, D1S551, and D1S2630) that were completely stable in H7 ES cells over passages. In addition, H1 ES cell showed instability in eight markers related to genes expressed during early differentiation (D2S134, D3S1583, G60405, D11S909, D18S63, DXS981, D17S2180, and DXS1208). In contrast, H7 ES cells showed instability in three different markers related to differentiation (D16S3091, D1S468, and D12S1682). Both ES cell lines showed instability in the differentiation marker DXS1208, but the difference did not reach significance. Statistically significant differences were observed in two pluripotency related markers (OCT4 and D1S551) and three differentiation related markers (G60405, D18S63, and D1S468). D1S551, D18S63, and D1S468 markers showed higher mean values of mutation frequencies at a significant level (p<0.05) compared with the other unstable markers analyzed (Figure 4 and Table 2). We suggest that the presence of genomic instability in markers located near to these specific pluripotency or differentiation genes could be a signal of gene expression changes that induce adaptation or differentiation of the ES cell during long term cultures and multiple passages.
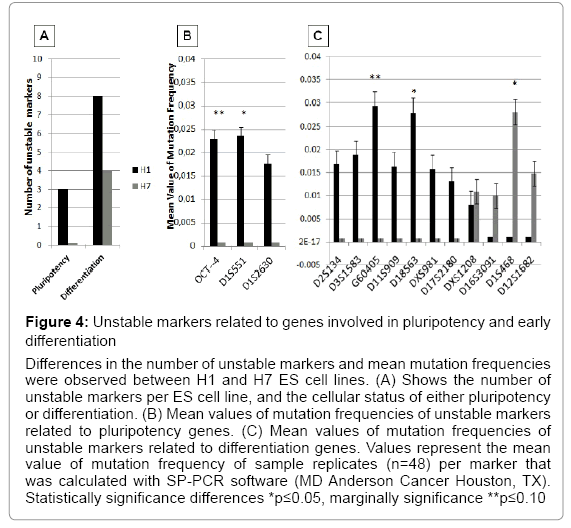
Figure 4: Unstable markers related to genes involved in pluripotency and early differentiation
Differences in the number of unstable markers and mean mutation frequencies were observed between H1 and H7 ES cell lines. (A) Shows the number of unstable markers per ES cell line, and the cellular status of either pluripotency or differentiation. (B) Mean values of mutation frequencies of unstable markers related to pluripotency genes. (C) Mean values of mutation frequencies of unstable markers related to differentiation genes. Values represent the mean value of mutation frequency of sample replicates (n=48) per marker that was calculated with SP-PCR software (MD Anderson Cancer Houston, TX). Statistically significance differences *p≤0.05, marginally significance **p≤0.10
| ES cells | Passage | Oct4 | D1S551 | G60405 | D18S63 | D1S468 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | n | m | f | n | m | f | n | m | f | n | m | f | n | m | f | |
| 28 | 46 | 2 | 0.031 | 54 | 0 | 0 | 33 | 0 | 0 | 37 | 0 | 0 | 45 | 0 | 0 | |
| H1 | 42 | 37 | 0 | 0 | 56 | 2 | 0.024 | 48 | 2 | 0.029 | 37 | 2 | 0.028 | 29 | 0 | 0 |
| 82 | 35 | 1 | 0.016 | 63 | 0 | 0 | 32 | 0 | 0 | 41 | 0 | 0 | 33 | 0 | 0 | |
| 27 | 33 | 0 | 0 | 48 | 0 | 0 | 33 | 0 | 0 | 63 | 0 | 0 | 55 | 0 | 0 | |
| H7 | 42 | 27 | 0 | 0 | 74 | 0 | 0 | 25 | 0 | 0 | 34 | 0 | 0 | 64 | 3 | 0.028 |
| 78 | 75 | 0 | 0 | 74 | 0 | 0 | 35 | 0 | 0 | 42 | 0 | 0 | 61 | 0 | 0 | |
| p-value | 0.06 | 0.046 | 0.077 | 0.036 | 0.05 |
Table 2: Mutation frequencies of five single tandem repeat markers located near genes related to pluripotency and differentiation Number of normal alleles (n), number of mutated alleles (m), and mean value of mutation frequency (f) calculated by SP-PCR software (MD Anderson Cancer Houston, TX). p-values ≤0.05 are in bold, p-value ≤0.10 in italic.
Epigenetic changes that occur during embryonic stem cell in vitro culture could result from genomic instability
Imprinting, chromatin assembly, and methylation are essential epigenetic mechanisms that modulate ES cell maintenance [23-25]. We found significant differences in ES cell genomic instability following passages. H1 ES cells showed instability in three markers (D22S447, D6S2252, and D10S529) and H7 ES cells in two markers (D10S529 and HISTHB2) that were related to chromatin assembly (Figure 5 and Table 3). All four chromatin assembly markers were significantly unstable. D22S447 and D6S2252 showed higher mean values of mutation frequencies at significant levels (p<0.05). Instability of the HISTHB2 marker was highly statistically significant in the H7 ES cells (p<0.001). H1 and H7 ES cells showed significant instability differences in the D10S529 marker (p<0.03) (Figure 5). Additionally, unstable markers for imprinting genes were determined. A single tandem repeat in the promoter of GRB10 imprinting gene was found to be unstable in both H1 and H7 ES cells, with a significant difference between them (p=0.026) (Figure 6) (Table 4). H7 ES cells also showed high instability in two additional markers (D2S144 and IGF2-PROM), whereas H1 ES cells were stable for these markers. D2S144 was significantly unstable compared with the IGF2-promoter marker that showed less significance (p=0.04 and p=0.08 respectively) (Figure 6 and Table 4). These findings showing instability of markers located near genes that participate in epigenetic modifications support the idea that genomic instability could be essential to generating epigenetic modifications during ES cell maintenance in vitro.
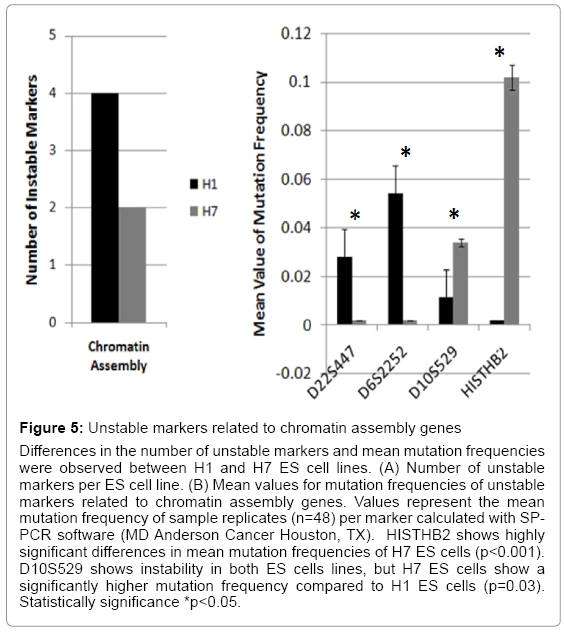
Figure 5: Unstable markers related to chromatin assembly genes
Differences in the number of unstable markers and mean mutation frequencies were observed between H1 and H7 ES cell lines. (A) Number of unstable markers per ES cell line. (B) Mean values for mutation frequencies of unstable markers related to chromatin assembly genes. Values represent the mean mutation frequency of sample replicates (n=48) per marker calculated with SPPCR software (MD Anderson Cancer Houston, TX). HISTHB2 shows highly significant differences in mean mutation frequencies of H7 ES cells (p<0.001). D10S529 shows instability in both ES cells lines, but H7 ES cells show a significantly higher mutation frequency compared to H1 ES cells (p=0.03). Statistically significance *p<0.05.
| ES cells | PassageNumber | D6S2252 | H1STHB2 | D10S529 | D22S447 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | m | f | n | m | f | n | m | f | n | m | f | ||
| 28 | 67 | 0 | 0 | 47 | 0 | 0 | 47 | 0 | 0 | 46 | 0 | 0 | |
| H1 | 42 | 41 | 3 | 0.054 | 38 | 0 | 0 | 39 | 0 | 0 | 37 | 2 | 0.028 |
| 82 | 49 | 0 | 0 | 40 | 0 | 0 | 40 | 1 | 0.012 | 45 | 0 | 0 | |
| 27 | 33 | 0 | 0 | 35 | 7 | 0.102 | 40 | 3 | 0.034 | 53 | 0 | 0 | |
| H7 | 42 | 42 | 0 | 0 | 34 | 0 | 0 | 40 | 0 | 0 | 38 | 0 | 0 |
| 78 | 63 | 0 | 0 | 37 | 0 | 0 | 36 | 0 | 0 | 68 | 0 | 0 | |
| p-value | 0.018 | <0.001 | 0.03 | 0.05 |
Table 3: Mutation frequencies of three single tandem repeat markers located near genes related to chromatin assembly
Number of normal alleles (n), number of mutated alleles (m), and mean value of mutation frequency (f) calculated by SP-PCR software (MD Anderson Cancer Houston, TX). p-value ≤ 0.05 are in bold
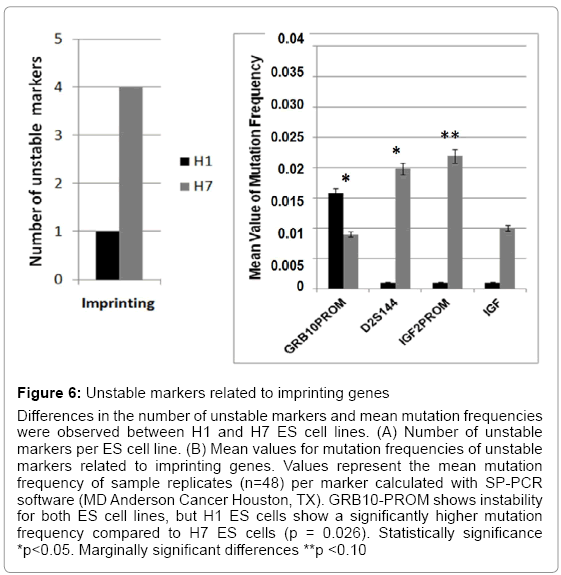
Figure 6: Unstable markers related to imprinting genes
Differences in the number of unstable markers and mean mutation frequencies were observed between H1 and H7 ES cell lines. (A) Number of unstable markers per ES cell line. (B) Mean values for mutation frequencies of unstable markers related to imprinting genes. Values represent the mean mutation frequency of sample replicates (n=48) per marker calculated with SP-PCR software (MD Anderson Cancer Houston, TX). GRB10-PROM shows instability for both ES cell lines, but H1 ES cells show a significantly higher mutation frequency compared to H7 ES cells (p = 0.026). Statistically significance *p<0.05. Marginally significant differences **p <0.10
| ES cells | PassageNumber | GRB10-PROM | D2S144 | IGF2-PROM | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | m | f | n | m | f | n | m | f | ||
| 28 | 44 | 0 | 0 | 45 | 0 | 0 | 66 | 0 | 0 | |
| H1 | 42 | 45 | 1 | 0.016 | 28 | 0 | 0 | 54 | 0 | 0 |
| 82 | 40 | 0 | 0 | 34 | 0 | 0 | 65 | 0 | 0 | |
| 27 | 35 | 0 | 0 | 37 | 0 | 0 | 53 | 0 | 0 | |
| H7 | 42 | 71 | 1 | 0.009 | 41 | 1 | 0.02 | 58 | 2 | 0.022 |
| 78 | 63 | 0 | 0 | 64 | 0 | 0 | 45 | 0 | 0 | |
| p-value | 0.026 | 0.04 | 0.08 | |||||||
Table 4: Mutation frequencies of three single tandem repeat markers located near genes related to imprinting genes. Number of normal alleles (n), number of mutated alleles (m), and mean value of mutation frequency (f) calculated with SP-PCR software (MD Anderson Cancer Houston, TX). p-values ≤ 0.05 are in bold, p-value ≤ 0.10 in italic.
Embryonic stem cells have the capacity for unlimited stem cell proliferation and the ability to differentiate into all the cell lineages derived from the three germinal layers. Questions about the molecular signals of pluripotency and self-renewal maintenance in vitro are still unanswered and are the key to clinical ES cell applications. We evaluated microsatellite marker instability in sequences located near genes known to be responsible for pluripotency and cell differentiation characteristics of ES cells.
Accumulation of DNA damage is observed during cellular stress responses. ES cells in long term cultures have shown genomic instability in response to environmental changes in the form of chromosomal abnormalities after more than 100 passages during in vitro maintenance [9-12]. Genomic instability in single tandem repeats create frame-shift mutations, and enhancer, or repressor modifications that could effect gene expression changes affecting cellular processes. This has been explored widely in tumorigenesis studies [26-30].
ES cells and tumors have common molecular pathways that maintain their cellular characteristics and functions [31-35]. Instability of a single tandem repeat located downstream or upstream of specific pluripotency and self-renewal genes is a reliable tool to characterize genomic stability during ES cell culture in vitro. It can be a potential biomarker to predict and evaluate pluripotency loss and uncontrolled cell differentiation processes during ES cell maintenance.
Our data suggest that instability in pluripotency and differentiation markers is a signal of balance between culture adaptation of ES cells and the differentiation process that is observed as morphological characteristics and genetic stability. H1 colonies became more irregular than did H7 colonies through culture passages. Colony irregularities are morphological signs of differentiation during cell culture and could be related to the DNA instability found in specific markers located near essential genes responsible for optimal ES cell functions. ES cells show low instability during early passages when compared to the mean frequencies of instability during middle and late passages. Several reports suggest that late passages significantly increase the frequency of chromosomal instability due to environmental signals from the in vitro system used to maintain ES cell lines in culture [9-12]. Our results support the idea that ES cell lines exhibit different adaptation processes, seen as genomic instability in early and middle passages as a part of cell adaptation in vitro. However, during later passages, chromosomal instability that enables maintenance of ES cell pluripotency occurs in some stem cell lines. Some studies report that the H1 ES cell line showed trisomy in chromosomes 12 and 17 at 144 passages [10,12,35]. In contrast, H7 ES cell line showed trisomy in chromosome 20 and translocation between chromosome 6 and 17 at passage 209 [10,12,35]. Apparently, chromosomal instability and single tandem repeat instability occur by independent processes that happen during longterm ES cell culture. H1 and H7 ES cell lines showed high rates of single tandem repeat instability during passages 27-28 and 42, but instability frequencies decreased at late passages (78-82 respectively) (Figure 3).
The key findings that emerged from this data included the failure of ES Cells to maintain pluripotency, tendency to differentiate, and epigenetic changes over passages. We identified twelve unstable markers localized near pluripotency, differentiation, chromatin assembly, and imprinting genes that play important roles during early embryogenesis. These genes are involved in specific cell signals that determine genetic and epigenetic modifications relevant to the ES cell: DNA transcription, cell cycle, cell differentiation, tissue specification, apoptosis, and DNA repair.
ES cell genes for pluripotency and self-renewal are actively expressed and are responsible for maintaining all characteristics of the ES cell. When genomic instability occurs around these specific genes, it could lead to loss of pluripotency and self-renewal in the ES cells. We found two unstable pluripotency markers in H1 ES cells; OCT4 and D1S551. OCT4 (POU class 5 homeobox 1) is a transcription factor that plays a role in embryonic development and has been identified as an important gene for ES cell pluripotency [36-38]. OCT4 is part of the ES cell gene network that regulates pluripotency by transcription regulation. OCT4, NANOG, and SOX2 are transcription factors that regulate themselves and bind common target developmental genes important for ES cell maintenance and embryonic development [37,39- 41]. D1S551 is located near a regulator of a G- protein signaling gene. G-proteins are involved in many cell signaling pathways [42-45]. In mouse ES cells, G- protein signaling is present during early neurogenesis and provides control of neuronal differentiation. Studies in mice and rats demonstrated that G- protein is a modulator of calcium channels, and gamma-aminobutyric acid (GABA), and opioid receptors [42,46].
Several reports have shown how gene expression changes occur during ES cell culture passages, but the exact mechanism is not clear [7,22]. Accumulation of DNA damage creates changes in gene expression that induce decline in cell function and loss of the cell’s integrity over time [3,7,8,22]. Long term culture and passages generate oxidative stress that is a source of DNA damage, apoptosis, and cell cycle defects [47-50]. For example, mouse ES cells, after exposure to ionizing radiation, show DNA damage that induces fibroblast cell differentiation [51,52]. Our results, show genomic instability could be a signal of gene expression deregulation. Early embryonic differentiation genes showed genomic instability in H1 ES cells over multiple passages. H1 ES cells cannot completely maintain pluripotency, whereas H7 ES cells can. Differentiation markers that showed instability in H1 ES cells were D2S134, D11S909, D18S63, and DXS981. Interestingly, these are specific markers located near to genes expressed during early embryonic neuroectoderm specialization [53-59]. D17S2180 and DXS1208 are related to endoderm and mesoderm specialization genes, respectively [60-64] (Table 5). In comparison to H7 ES cell unstable markers, D16S3091 is related to early mesoderm gene differentiation, D1S468 is a gene that promotes apoptosis, and D12S1682 is both an endoderm and mesoderm differentiation gene [65-67] (Table 5).
| Name Marker | Gene Symbol | Gene Name | Gene Function | References |
|---|---|---|---|---|
| Oct4 | POU2F1 | POU class 2 homeobox 1 | Developmental transcription factor | [36,72] |
| D1S551 | RGS4 | Regulator of G-protein signalling 4 | Signal transduction regulator | [42,43] |
| D1S2630 | POU2F1 | POU class 2 homeobox 1 | Developmental transcription factor | [38,40,41] |
| D2S134 | ME1S1 | Meishomeobox 1 | Embryo development | [57,59] |
| D3S1583 | RARB | Retinoic acid receptor, bet | Embryo development | |
| G60405 | ERCC6 | Excision repair cross-complementing group 6 | DNA repair | |
| D11S909 | ST5 | suppression of tumorigenicity 5 | Tumor suppressor | |
| D18S63 | TG1F1 | TGFB-induced factor homeobox 1 | growth factor activity | [54,56] |
| DXS981 | SPG16 | spastic paraplegia | Developmental gene | [53] |
| D17S2180 | HOXB5 | homeobox B5 | Developmental transcription factor | [60,62] |
| DXS1208 | HSPB1 | Heat shock thermic protein | Developmental transcription factor | [64] |
| D16S3091 | CDF13 | cadherin 13, H-cadherin (heart) | Growth factor activity |
Table 5: List of unstable markers Summary of characteristics of genes located in close proximity to unstable markers involved in embryonic development. Twelve markers showed statistically significant instability frequencies
Genomic instability is a multistep process that involves genetic and epigenetic modifications that induce opposite effects on the status of ES cell pluripotency. Epigenetic changes such as chromatin assembly, imprinting, and methylation are responsible for determining transcriptional patterns dependent upon the cell stage. Imprinting is a switch for gene transcription that ensures cell proliferation, development, and tissue specific functions [50,66,68- 71]. Developmental genes for ES cells have a specific pattern of histone modifications that determine the status of activation of specific genes involved in embryonic development and cell fate during differentiation by de novo methylation. For example, the OCT4 gene is unmethylated during pluripotency by bivalent histone modifications to ensure cell proliferation and development. However, OCT4 is completely repressed when cell differentiation occurs [72-74]. ES cell lines in vitro fail to maintain a specific epigenetic pattern, inducing changes in the cellular status that leads to loss of ES cell pluripotency over time [23,25,50,75]. In our study, H1 and H7 ES cells showed significant differences of instability in markers that were located next to chromatin assembly and imprinting genes across time. Genomic instability was observed in markers such as D22447, D6S2252, HISTHB2, and D10 S529, all of which were located near to genes that code for histone proteins. The D6S2252 marker is located next to HIST1H2AH (linker histone H1), which interacts with the DNA between nucleosomes and is responsible for chromatin compaction [33,76,77]. D10S529 is a marker for a variant histone, H2AFY2 that contributes to the inactivation of the X chromosome [78-80]. In zebra fish embryos, it has been observed that H2AFY2 is involved in the activation of neuronal differentiation genes such as the homeobox A1 gene (HOXA), which encodes a DNA-binding transcription factor to control gene expression during embryonic development and cell differentiation [79]. D22S447 is a histone cell cycle regulator A (HIRA) that is a homolog of Saccharomyces cerevisiae histone. HIRA is responsible for controlling cell growth by regulation of cell cycle genes [81]. Taken together, our results suggest that instability in these markers could be the signals that induce X chromosome inactivation, ES cell growth, and differentiation through changes in expression of developmental and differentiation genes over multiple passages. Additionally, imprinting markers showed instability and are involved in the embryonic methylation process. D2S144 is a marker for the DNA (cytosine-5-)-methyltransferase 3 alpha gene (DNMT3A) that is responsible for epigenetic modification of de novo DNA methylation important for embryonic development, differentiation, imprinting, and X-chromosome inactivation [82,83]. Other unstable markers are located next to the promoter region of imprinting genes, such as GRB10 and IGF2, which are imprinted in a tissue specific manner. These results confirm that H1 and H7 ES cells have a constant and actively regulated process across passages that control genetic and epigenetic outcomes to ensure ES cell growth, maintenance of cell feature characteristics or cell differentiation in vitro.
In conclusion, our findings indicate that maintenance of ES cell genetic and epigenetic characteristics is compromised by the loss of DNA integrity in tandem repeat sequences that flank specific genes that are responsible for the pluripotency and self- renewal of ES cell maintenance, cell fate during differentiation, chromatin assembly, imprinting, and methylation. From our data, we can support the idea that genomic instability could be responsible for genetic and epigenetic imbalances originating in long term ES cell cultures. The exact signals that coordinate this process are complex and not completely known. Even so, our data support our hypothesis that instability in repetitive sequences located close to specific genes could be the signal for adaptation or differentiation of ES cells in culture passages over time. Future studies could be focus in evaluate the level of expression and methylation of these candidate proximal genes.
Furthermore, our results identify biomarkers that could be part of an ES cell characterization process that evaluates genomic integrity through in vitro maintenance procedures. Understanding the role of genomic instability in ES cell maintenance could lead to the origin of an accurate approach for the safety and reliability needed in regenerative medical applications of human ES cells.
We would like to thank the National Stem Cell Bank for providing the H1 (WA01) and H7 (WA07) human embryonic stem cell lines. Also, we extend our thanks to Dr. K. Sue O’Shea and Crystal Pacut at the Consortium for Stem Cell Therapies at the University of Michigan for providing the DNA samples from late passage of H1 and H7 ES cell lines. We would like to thank Dr. Karen Coats, Dr. Janet Donaldson, and Kortney Wilkinson for manuscript editing. This research was funded by the Department of Biological Sciences; Office of Institutional Research; Office of the Graduate School, and the College of Art and Sciences at Mississippi State University.