Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 5
The genus Helicobacter comprises several species of both gastric and enterohepatic intestinal bacteria. H. pylori, the type species of the genus, is associated with gastritis, peptic ulcer and gastric cancer in humans. H. pylori genome has a high number of restriction and modification (R-M) systems and their diversity is useful for strain typing. To analyse if such a high number of expressed methyltransferases is a characteristic of the genus Helicobacter, the genomic methylation of five non-pylori Helicobacter spp. (H. canadensis, H. canis, H. felis, H. mustelae and H. pullorum) was determined. The results revealed that the number of R-M systems among nonpylori Helicobacter spp. is smaller than those observed among a group of 221 H. pylori strains (p<0,001), but is greater than those observed for the mean of all bacteria sequenced genomes (p=0,005). 16S ribosomal RNA analysis of H. pylori sequenced strains and five non-pylori Helicobacter spp. clearly isolate H. pylori species. Surprisingly, the analysis of the genomic methylation status by MCRM algorithm performs similarly. This suggests that R-M systems do not appear to be spread in a miscellaneous manner, once even that these genes may be subjected to acquisition and loss; their expression still allows discriminating among Helicobacter spp.
Keywords: MCRM algorithm, Genomic methylation, Helicobacter spp., Bioinformatics, Restriction and modification, ribosomal RNA.
MCRM: Minimum Common Restriction and Modification; REase: Restriction Endonuclease; MTase: Methyltransferase; RM: Restriction and Modification.
Helicobacter pylori colonizes the stomach of about half of the human population and is associated with several disease outcomes, like gastritis, peptic ulcer and gastric cancer (Dunn et al., 1997; Kusters et al., 2006). Other similar spiral bacteria have been isolated from several animals, like cat, dog or mice, among others. Helicobacter species can be subdivided into gastric Helicobacter species and enterohepatic (nongastric) Helicobacter species. The two lineages demonstrate a high level of organ specificity, such that gastric Helicobacter spp. in general does not colonize the intestine or liver, and vice versa (Kusters et al., 2006).
The genomic DNA of H. pylori is characterised by the presence of a high unusual number of restriction and modification (R-M) system (Nobusato et al., 2000; Lin et al., 2001; Takata et al., 2002). The type II R-M systems are composed by least two genes: one coding for a restriction endonuclease (REase) that recognizes a specific DNA sequence and cuts both strands; and other gene coding for a cognate MTase that methylates the same DNA sequence, thus protecting the genomic DNA from being cleaved by the companion REase (Roberts et al., 2003). Type II R-M systems have been referred as selfish genetic elements, because the descendants of cells that had lost these genes appeared unable to modify a sufficient number of recognition sites in their chromosomes to protect them from lethal attack by the remaining restriction enzyme molecules (Naito et al., 1995).
Recently we have demonstrated that the diversity of RM systems among H. pylori strains is high enough to be used as a typing method (Vale and Vitor, 2007). Cluster analysis by conventional methods does not consider the propensity for R-M systems conservation after acquisition, due to the “selfish behavior” (Naito et al., 1995). Considering this we have recently developed a new clustering algorithm [Minimum Common Restriction Modification (MCRM) algorithm] that takes into account the pressure of REases on MTases, and that is based on the hypothesis that each strain evolves by acquiring new RM systems without loosing acquired RM systems (Vale et al., 2008). In this algorithm it is considered that: i) the strain with less RM systems is the one that has the core set of the most abundant R-M systems expressed among the typed strains; ii) these core set of RM systems was the first to be acquired by H. pylori, so that they exhibit a large dissemination (expression) among several daughter strains (Vale et al., 2008). MCRM analysis of the genomic methylation data from H. pylori strains isolated from different geographic revealed a clustering according to strain’s continent of origin (Vale et al., 2008), which is in agreement with H. pylori coevolution with its human host (Covacci et al., 1999; Linz et al., 2007; Vale et al., 2008). This observation led to the suggestion that R-M systems may trace H. pylori geographic distribution and, by default also its human host migrations (Vale et al., 2008).
In this study it was investigated if non-pylori Helicobacter strains also have a high number expressed MTases and, if the genomic methylation status followed be MCRM algorithm analysis permitted to discriminate between H. pylori and non-pylori Helicobacter spp. (H. canadensis, H. canis, H. felis, H. mustelae and H. pullorum). Following, these results were compared with the phylogenetic analysis of 16S rRNA gene sequences for the same species. To our knowledge this is the first study that systematically analysis the diversity of expression of R-M systems in H. canadensis, H. canis, H. felis, H. mustelae and H. pullorum.
Helicobacter spp
H. pylori strains (26695 and J99) were cultured on H. pylori selective agar (Wilkins-Chalgren agar supplemented with 10% horse blood, vancomycin [10 mg liter-1], cefsulodin [5 mg liter-1], trimethoprim [5 mg liter-1], and cycloheximide [100 mg liter-1] [Biogerm, Porto, Portugal]) and incubated at 37°C for 48 h in an anaerobic jar (Oxoid, UK; or BBL, USA) with a gas generator system (CampyGen; Oxoid, UK) (Megraud, 1996). Non-pylori Helicobacter spp. were cultured on Muller Hinton agar (Oxoid, UK) supplemented with 10% (v/v) defibrinated horse blood (Probiologica, Portugal) and incubated in similar conditions. Genomic DNA was extracted by standard methods.
Helicobacter spp. R-M Systems Diversity
To evaluate the expression of the cognate methyltransferase, the genomic DNA was digested with 27 REases [AciI, AseI, BseRI, BssHII, BstUI, DdeI, DpnI, DpnII, DraI, EagI, FauI, Fnu4HI, FokI, HaeIII, HhaI, Hpy188I, Hpy188III, Hpy99I, HpyCH4III, HpyCH4IV, HpyCH4V, MspI, NaeI, NlaIII, Sau96I, ScrFI, and TaqI (New England Biolabs, USA)]. The results were coded as“0” for digestion observed (DNA is unmethylated), and “1” for absence of digestion, suggesting an active methyltransferase (Vale and Vitor, 2007).
Genomic Methylation Status Comparison
The mean number of active MTases on non-pylori Helicobacter spp. was compared with: i) the mean number of expressed MTases of 221 H. pylori tested by us; ii) the mean number of M genes predicted by REBASE for all sequenced bacteria (Roberts et al., 2007). The Kruskal- Wallis test was performed using the statistical package SPSS v.15 (SPSS Inc., Chicago, IL).
Ribosomal RNA Alignment
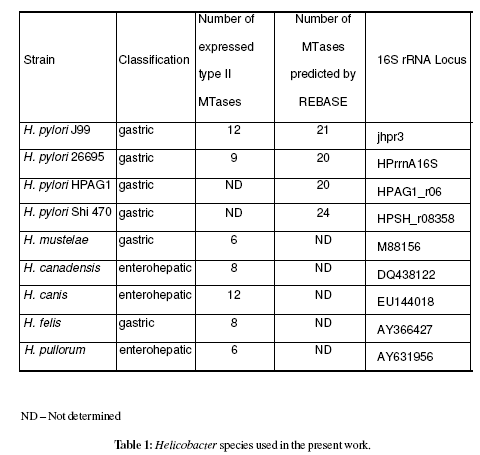
16S rRNA sequences available on public data bases from the Helicobacter spp. (table 1) were aligned using ClustalW, producing a cladogram (Chenna et al., 2003).
MCRM Clustering
Several dendrograms were produced using MCRM algorithm after genomic methylation analysis of 7 Helicobacter spp. Most of the dendrograms produced by MCRM algorithm are indeed similar, but it is possible that distinct dendrograms are generated as different choices of strain or R-M system at ties may result in different clustering. Thus, 10 different dendrograms were produced from the same data in order to increase the confidence on the clustering results.
Helicobacter spp. Genomic Methylation
After genomic DNA hydrolysis with the selected REases it was observed that among non-pylori Helicobacter spp. the mean number of expressed MTases was 8 (SD=2.4). A similar analysis for 221 H. pylori strains (Vale et al. unpublished results) revealed a mean of 17 active MTases (SD=3.4). The mean number of genes coding for methyltransferases for the overall 862 sequenced genomes is 4.2 (SD=5.0) [data from REBASE (Roberts et al., 2007)]. Table 1 resumes the number of active methyltransferases in tested Helicobacter spp.
A significant statistical difference between mean number of active MTases from non-pylori Helicobacter spp. and H. pylori (p<0,001) and also between non-pylori Helicobacter spp. and the overall sequenced genomes (p=0,005) was verified. Present study results showed that the number of expressed MTases in a decreasing order by organism, or group of organisms, was: H. pylori, non-pylori Helicobacter and all sequenced bacteria available at REBASE (Roberts et al., 2007).
Helicobacter spp. 16S rRNA Cladogram
Construction of a cladogram after multiple sequence alignment using ClustalW (Chenna et al., 2003) 16S rRNA sequences (from Helicobacter spp.) available on public databases clearly isolated H. pylori species (Figure 1). As expected the H. pylori sequenced strains (26695, J99, HPAG1 and Shi470) presented a similarity level =98%, and clustered together (>97% defines a species). Moreover, Helicobacter gastric and enterohepatic species appear to be in different clusters, according to previous work (Dewhirst et al., 2005). The similarity levels among Helicobacter spp. based on 16S rRNA is presented in table 2.

Figure 1: Cladogram based on 16S rRNA of Helicobacter species used
MCRM Clustering Analysis
After genomic DNA hydrolysis with the selected REases the codified data were analysed using MCRM algorithm (Vale et al., 2008). The Simpson index of diversity, which reflects the capacity of the method to distinguish unrelated strains, was 100% (Hunter and Gaston, 1988). The produced dendrogram is present in Figure 2. Surprisingly, this analysis based on the genomic methylation status clearly isolated H. pylori species from non-pylori Helicobacter spp., as it was observed when the analysis focuses on the 16S rRNA. Out of 10 produced dendrograms, 60% cluster H. pylori together and 40% of the dendrograms also discriminate between H. pylori and non-pylori Helicobacter spp. All of these last mentioned dendrograms gathered H. pylori strains (data not shown). H. pylori was discriminated from non-pylori Helicobacter spp. at k/nM=0.04 (where, k=1 and nM=27, i.e. one MTase common to all Helicobacter species used). This MTase common to all tested Helicobacter spp. was M.NaeI (table 3).
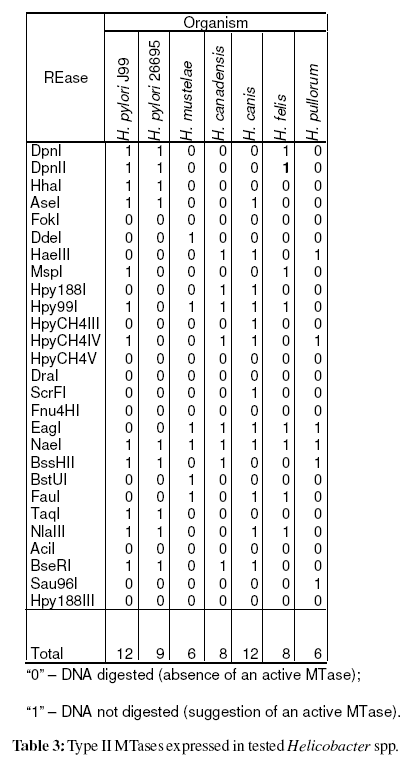

Figure 2: MCRM algorithm clustering of Helicobacter species used. Similarity represented by K/nM, where K is the number of R-M systems common to all tested strains and nM the total number of R-M systems screened.
When comparing the number of MTases expressed in each Helicobacter spp. it was verified that H. pylori expresses a number of MTases higher than tested non-pylori Helicobacter spp. (p<0,001) and, that non-pylori Helicobacter spp. expresses a number higher than all sequenced bacteria analysed by REBASE (p=0,005). This analysis reveals that the increased number of MTases genes expressed is probably a characteristic of Helicobacter genus and not only of H. pylori. To our knowledge the evaluation of the expressed MTases in non-pylori Helicobacter spp. has only been referred for the sequenced genomes of H. acinonychis Sheeba (Eppinger et al., 2006) and H. hepaticus ATCC 51449 (Suerbaum et al., 2003) with 29 and 8 M genes, according to REBASE (Roberts et al., 2007), respectively. Besides this analysis only the sequence GATC has been screened for methylation in H. mustelae (Edmonds et al., 1992). Present study and Edmonds et al. study (Edmonds et al., 1992) found that the GATC methylation is absent in H. mustelae (table 3).
The only MTase that it was found to be expressed among all tested Helicobacter spp. is M.NaeI (table 3). Previously we have reported that this MTase is probably conserved in all H. pylori strains (Vale and Vitor, 2007; Vale et al., 2008). In order to confirm if this MTase is common to the Helicobacter genus, other non-pylori Helicobacter spp. should be included in the study, and for each species several stains should be characterized.
Analysis of 16S rRNA gene sequences has become the primary method for determining prokaryotic phylogeny, which is the current basis for prokaryotic systematics. Although it has been described that Helicobacter is susceptible to horizontal transfer of 16S rRNA gene so that it can be misleading in Helicobacter spp. identification (Vandamme et al., 2000; Dewhirst et al., 2005), the 16S rRNA cladogram clearly discriminate the Helicobacter spp. used in the present study as expected. Moreover, figure 1 presents gastric Helicobacter spp. and enterohepatic Helicobacter spp. in different clusters, as described elsewhere (Dewhirst et al., 2005). However, the cladogram obtained from 60 kDa heat-shock protein (HSP60), referred as better marker for Helicobacter species phylogeny (Mikkonen et al., 2004) was similar to the one obtained with 16S rRNA (data not shown).
A surprising result was the capacity of the genomic methylation and of MCRM algorithm to cluster separately gastric Helicobacter spp. and enterohepatic Helicobacter spp. in 40% of the produced dendrograms. Also this methodology discriminate H. pylori from non-pylori Helicobacter spp. in 60% of the produced dendrograms. When genomic methylation and MCRM analysis is compared with the current phylogeny approach the results are remarkably similar. The genomic methylation status appears to be an interesting new tool to characterize genomes with a high number of MTases expressed. It has been described that R-M systems are subject to horizontal transfer (Jeltsch and Pingoud, 1996; Gressmann et al., 2005). The separation between gastric Helicobacter spp. and enterohepatic Helicobacter spp. suggests that probably theses species, which have a high level of organ specificity (Kusters et al., 2006) may have access to different sets of R-M systems through horizontal gene transfer. The horizontal gene transfer might occur only in ideal conditions provided by the specific tissue environment characteristic of each species. This could justify the presence of gastric Helicobacter spp. and enterohepatic Helicobacter spp. in different clusters. Similarly, H. pylori unique reservoir may justify the presence of the two tested strains in a different cluster. Finally, the results suggest that possibly some R-M systems are not as mobile as previously described, or are not available for horizontal transfer due to the isolation provided by the human (or animal) reservoir of each species, because the genomic methylation analysis permits to discriminate among Helicobacter spp. The R-M systems do not appear to be spread in a miscellaneous manner.
Indeed, a blast analysis (Zhang et al., 2000) of all methyltransferases predicted by REBASE (Roberts et al., 2007) for the recently sequenced H. pylori strain Shi470 clearly shows that most methyltransferases are identical to other H. pylori sequenced strains and to H. acinonychis strain Sheeba (table 4, supplementary material). It is clear from analysis of table 4 that most of the methyltransferases of the recent sequenced H. pylori strain Shi470 are identical to the other H. pylori sequenced strains and H. acinonychis strain Sheeba, but none is observed in H. hepaticus sequenced strain. We postulate that similarly to H. pylori, non-pylori Helicobacter may also present a high diversity of MTases expressed which could be used for strain typing, but this still needs to be confirmed with further investigation. R-M systems probably play an important role in Helicobacter genus biology that has not been ascertained, yet.
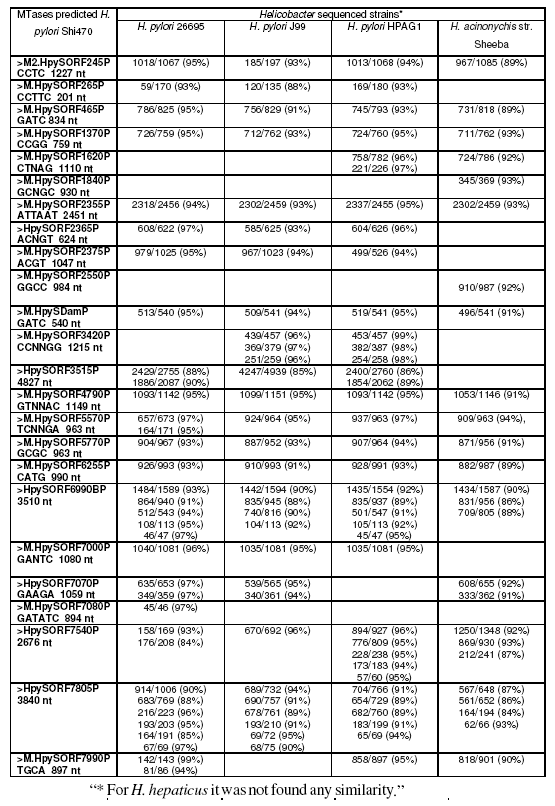
Table 4:. Blast results of DNA sequence of MTases from H. pylori strain Shi470 predicted by REBASE against all other Helicobacter spp. sequenced strains.
In conclusion, it was observed a high number of MTases expressed in non-pylori Helicobacter spp. as it was previously determined for H. pylori. The discrimination of Helicobacter species by the dendrogram produced with MCRM algorithm and by 16S rRNA alignment performed in a similar way. The results suggest that some R-M systems do not appear to be spread in a miscellaneous manner, once genomic methylation analysis allows discrimination among Helicobacter spp. Future work should include increasing the number of Helicobacter species analysed and also the number of tested strains from each species, in order to confirm present study results.
We thank Lurdes Monteiro, Francis Mégraud, Jay Solnick and, Nuno Azevedo for the Helicobacter spp. strains. This work was partially supported by New England Biolabs, Inc. (USA).