Journal of Depression and Anxiety
Open Access
ISSN: 2167-1044
ISSN: 2167-1044
Review Article - (2014) Volume 3, Issue 1
Treatment of Major Depressive Disorder (MDD), in many cases, offers less than optimal results, resulting in loss of function and remaining impairment. This is partially explained by the high prevalence of comorbid psychiatric illnesses that share common features, likely resulting in less than adequate treatment outcomes among patients receiving traditional psychiatric treatment. The term anhedonia does not sufficiently capture the complex and multifaceted reward-associated deficits seen in neuropsychiatric disorders. Deficits in reward-related processes can lead to behaviors that may appear to be the loss of interest or pleasure, and may preclude an individual from engaging in goal-oriented action. Investigators have identified a lifelong poor hedonic responsiveness, manifested as altered sensitivity to reinforcement, in patients with Attention Deficit Hyperactivity Disorder (ADHD), and suggest it as a common endophenotype for depression and the inattentive symptoms of ADHD. Research into the neurobiology of anhedonia has focused on the centers in the brain related to reward and motivation, and has identified the dopaminergic and associated circuits as playing a key role. Studies have shown that lesions in reward and motivation circuits result in anhedonia, and that decreased activation in the medial frontal cortex is believed to underlie abnormal positive affect processing in patients. In this paper, we propose that a specific phenotype of depression, which is more likely to be described as “treatment resistant” is associated with high risk of comorbidity with dysthymia, ADHD, and substance abuse, specifically cocaine abuse. We believe these patients experience a chronically lower hedonic tone that results in chronic anhedonia (as opposed to acute state anhedonia noted in a major depressive episode) related to unique pathophysiology and requiring treatment of the dopaminergic deficiency that characterizes its unique collection of comorbidities.
Keywords: Anhedonia; Attention deficit hyperactivity disorder; Cocaine; Comorbidity; Major depressive disorder, Dopamine; Dopaminergic neurons; Limbic system; Prefrontal cortex; Reward
Treatment of Major Depressive Disorder (MDD) in many cases, offers less than optimal results, resulting in loss of function and remaining impairment. In recent years, there has been an accumulation of evidence demonstrating substantial etiopathological heterogeneity and overlap both within and across a variety of psychiatric disorders including depression [1]. While initial research was conducted to gain insight into genetic risk, much of the current research has focused on attempting to develop more effective treatments. This has proven to be difficult, in part, due to the varying degree of etiological heterogeneity of symptoms associated with MDD. Moving beyond genetics, towards an understanding of specific neural pathways and distinct disruptions that may result in related, but different, symptomatic presentation, could lead to a more accurate appraisal and definition of disorders. Still, at present, there is no consensus on how to identify relevant and disease-specific pathology with the specific neural circuits given our current limited understanding of disease pathophysiology.
Hence, the purpose of this paper is to gain insight and to propose a common link between symptoms and related disorders in association with specific dysfunction of known neural circuits, in order to account for the increased likelihood of developing treatmentresistant depression in a subgroup of depressed patients. This paper will specifically attempt to present the question as to whether low hedonic tone is a risk factor for Attention Deficit Hyperactivity Disorder (ADHD) and for developing comorbid depression. Furthermore, this paper will present the hypothesis that low hedonic tone and associated specific neurobiologic correlates predict the presence of ADHD in MDD, thus who is at risk for poorer outcomes with selective Serotonin Reuptake Inhibitors (SSRIs).
Eleven key areas of the brain have been highlighted in part by their specific neurotransmitter projections of which alteration in neurotransmitter innervation has been hypothesized to create disruptions in information processing and consequently specific psychological symptoms [2]. Linking each of these functional areas is a network of neuron projections or neurotransmitter pathways from the brainstem. It is posited that when a neurotransmitter-carrying neuron projects into a specific area, it has the capacity to modulate the physiological processes of the associated circuit [2]. Thus these pathways form the substrates that “tune” the neurons within each circuit, thereby affecting the function of the specific brain areas they innervate. Furthermore, it is hypothesized that these neural circuits are linked together in functional loops allowing various regions of the brain to communicate chemically via neurotransmission. In some instances, for example in cortex-to-cortex circuits, cortical areas are “tuned” by bottom-up neurotransmission, whereas other cortical circuits including a variety of Cortico-Striatal-Thalamic-Cortical Circuits (CSTC), allow information to be sent downstream by way of top-down mediation [3]. Whenever there is malfunction or even hypofunction in these circuits, the effects are felt throughout the loop and thus may mediate specific psychological symptoms.
This model, when applied to depression, illuminates the complex interaction of the three monoaminergic neurotransmitters system comprised of serotonin (5-HT), Norepinephrine (NE), and Dopamine (DA), which act to modulate the information processing across a wide variety of neural circuits, any and all of which may be responsible for mediating various symptoms of depression [3]. Specifically, the basis of this hypothesis rests on the notion that the symptoms of a major depressive episode (MDE) are directly related to inefficient information processing in various neural circuits, which can be mapped topographically to specific brain regions. Stahl [3] posits that not only can each symptom be mapped to specific brain regions, but also the monoaminergic neurotransmitters that innervate the region (Figure 1). Moreover, Nemeroff [4] asserted that depression can be characterized specifically as a dysfunction of the limbic system-cerebrocortical circuits, all of which are modulated by the catecholaminergic monoamines.
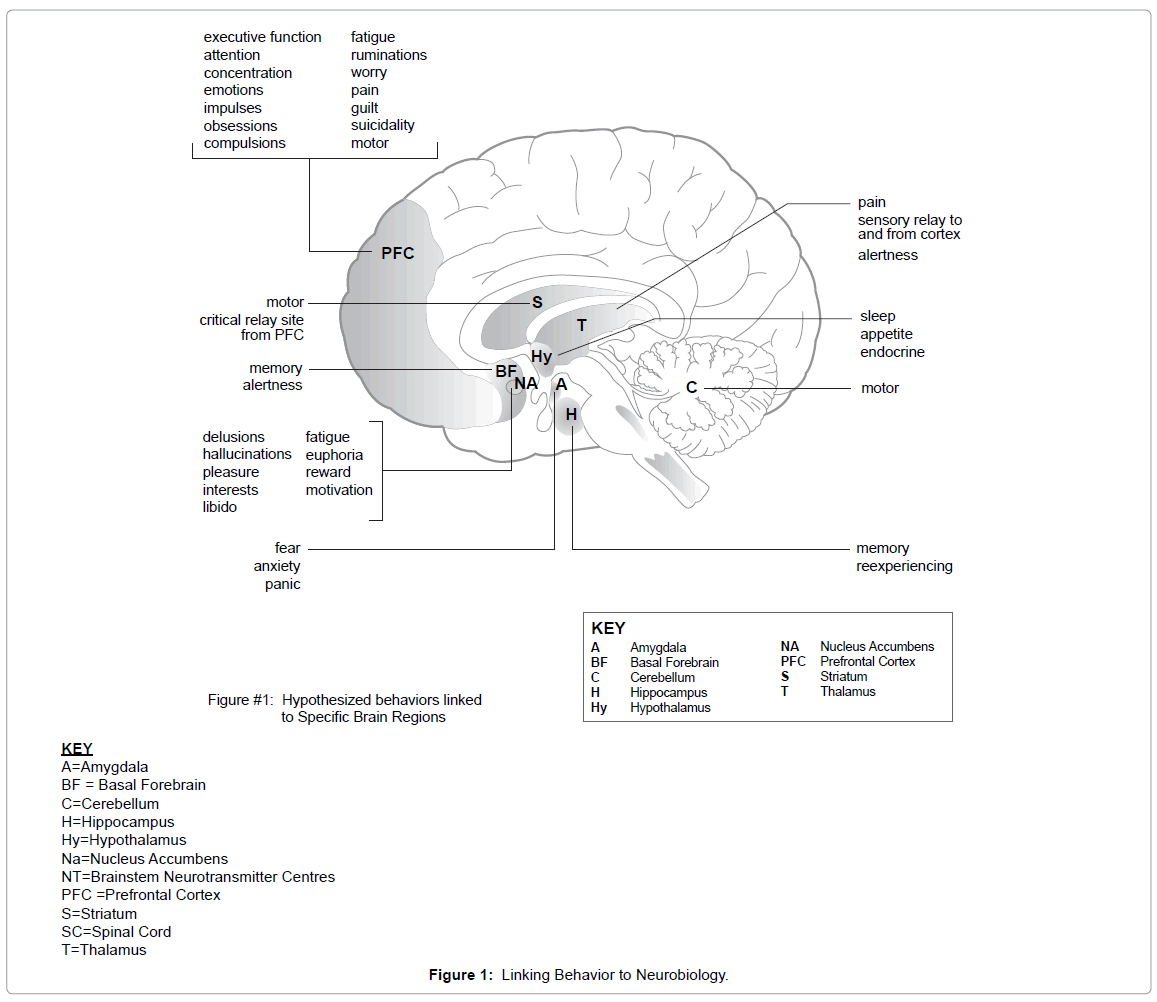
Figure 1: Linking Behavior to Neurobiology.
The monoamine hypothesis of depression postulates that depression is caused by decreased function of 5-HT, NE, or DA in the brain [5]. Early evidence for the role of monoamines in depression came from clinical observations that treatment with reserpine, a drug that depletes monoamine neurotransmitters, evoked clinical symptoms very similar to major depression in patients who were not formerly depressed [6,7]. Further evidence came from studies in which MDD patients in remission previously relapsed and became depressed following monoamine depletion [8]. In addition, monoamine depletion resulted in worsened mood in individuals with a family history of MDD and in drug-free remitted individuals with MDD.
While the monoamine hypothesis of depression is a widely accepted hypothesis that has governed the development and selection of pharmacological agents used to treat depression, it does not explain the apparent lack of efficacy in a relatively large subset of patient who continue to suffer with specific residual symptoms including lack of interest or pleasure and who often exhibit lifespan comborbid psychological disorders.
Most individuals with depression experience psychiatric comorbidity, according to the National Comorbidity Survey Replication (NCSR) [9]. Overall, 59.2% of respondents had comorbid anxiety disorders, 24.0% had comorbid substance use disorders, and 30.0% had comorbid impulse control disorders [10]. Among respondents with ADHD, there was significant comorbidity with a wide range of DSMIV disorders, with odds ratios of 2.7–7.5 for mood disorders, 1.5–5.5 for anxiety disorders, 1.5–7.9 for substance use disorders, and 3.7 for intermittent explosive disorder [9].
It is believed that the high comorbidity of MDD and substance use is due to common dysregulation in frontal-limbic systems associated with stress and reward pathways [11]. Evidence from neuroendocrine and neuroimaging studies indicate similar alterations in MDD and substance use, including decreased frontal metabolism [12-14], reduction in DA D2 receptors in frontal-striatal regions [14,15], reduced cell density and gray matter volume [13,16], hyperactivity in the amygdala [17-19], and hypoactivity in the anterior cingulate cortex [18,20,21] compared to healthy subjects.
Among the most studied of the illicit drugs that are abused is cocaine. Cocaine, like other stimulant substances, results in increased levels of DA within the nucleus accumbens, resulting in decreases in the brain stimulation reward threshold during use [22], and increased reward threshold during cocaine withdrawal [23]. During periods of abstinence, it has been reported that it is common for individuals to experience depression, fatigue (physical and mental), impaired concentration, and anxiety, often in association with withdrawal symptoms [24]. Dackis [25] asserted that cocaine withdrawal severity may result from cocaine-induced neurobiological changes and may identify patients with chronic hedonic dysregulation. Taken together this may explain the reaction of a subset of patients that, when questioned, note that cocaine increases focus, attention, and overall mood, thus suggesting these patients may be self-medicating with cocaine [26,27], and suggesting they are using cocaine (and other substances) in an attempt to manage their dysregulation. Often drug abusers will report the most positive effects on the first dose, no doubt as a result of pleasurable increases in phasic DA neuronal firing [28]. However, eventually the mesolimbic system will counteract this effect, with a compensatory tonic lowering of DA neuronal firing, resulting in withdrawal symptoms of feelings of intense sleepiness, anhedonia, and cravings for the agent between the doses [29]. Ultimately, the alterations in DA neuronal firing, results in a state of long lasting DA depletion, anxonal degeneration, which is directly associated with anhedonia and treatment-resistant depression [3].
When MDD is comorbid with ADHD, it is associated with a much more severe course, an earlier age of illness onset, more complex psychiatric comorbidities, decreased quality of life, greater burden of illness, greater illness complexity, lower social functioning, lower work productivity, and lower employment rates than patients with MDD alone [30,31]. In addition, individuals with MDD and ADHD experience higher levels of substance use and psychosocial impairment than disorder in isolation [32-35], suggesting a potential variant of MDD, and therefore explaining the treatment resistance in this population when treated with traditional SSRIs.
Neuroimaging studies have identified morphological and functional features common to both MDD and ADHD, including decreased brain volumes and altered activity of frontal lobe structures (and specifically the dorsolateral prefrontal and anterior cingulate cortex). Interestingly, these areas have been associated with attention regulation, behavior selection, and emotion [36,37]. Neurotransmitter studies have revealed abnormalities in DA and NE signaling in both MDD and ADHD, suggesting a potentially shared underlying pathophysiology, at least in some individuals [38-41].
Support for the dopaminergic theory of ADHD which states that ADHD results from DA deficits and therefore lowered activity in the frontal cortex and striatum [42], comes from a variety of pharmacological and imaging studies which have revealed hypoperfusion and lowered activations in those areas [43-46]. Interestingly, with treatment with methlphenidate, normalization of the hypoperfusion of prefrontal areas is associated with the corresponding improvement in ADHD symptoms [47-51].
These findings point to a key role for DA dysfunction in the pathophysiology of ADHD and its comorbidities. With evidence pointing to dysfunctional neuromodulation of the dorsolateral prefrontal cortex in relation to executive dysfunction in ADHD [52-56], it is not too much of a stretch to consider low hedonic tone as an affective correlate in ADHD explaining the lack of responsiveness to SSRIs in depression.
Several studies published in the literature report worse treatment outcomes for depressed patients with comorbid substance abuse. Worthington et al. [57] observed that the degree of alcohol consumption at baseline in non-substance-abusing outpatients with MDD was a significant predictor of poorer response to treatment with the SSRI fluoxetine. In a similar study, chronic depression at baseline and no more than moderate alcohol consumption during therapy were predictors of poor response to fluoxetine among adolescents with comorbid depression and substance use disorder [58]. Further to this point, compared to elderly patients with depression alone, elderly patients with lifetime alcohol use and current depression have a lower rate of recovery from depression 1 month after hospital discharge, more hospital readmissions for depression, and are more likely to experience lack of improvement in depression at follow-up [59]. The impact of alcohol use on the long-term outcomes of MDD were reported by Mueller et al. [60] who observed that lifetime alcohol problems decreased the likelihood of recovery from depression from 2.3% to 1.2% over 10 years.
Similar findings have been observed for the impact of drug abuse on outcomes among depressed patients. The Collaborative Care for Anxiety and Panic (CCAP) study demonstrated that combined Cognitive-Behavioral Therapy (CBT) and pharmacotherapy resulted in improvement in depression regardless of cannabis use status, while usual care resulted in improvement in depression only for those using cannabis less than monthly [61].
Further support of this hypothesis was demonstrated in the STAR*D study, where the adult patients with MDD treated with the SSRI, citalopram, who used both alcohol and drugs had significantly reduced rates of remission and significantly increased times to reach remission compared to patients with MDD alone [62]. In the Treatment of Resistant Depression in Adolescents (TORDIA) trial, (which was designed to identify predictors of relapse and remission in adolescents with SSRI-resistant depression), higher drug and alcohol use at 12 weeks of treatment predicted non-remission (OR=2.07, p=0.007) at the end of the 24-week study period [63]. Not all studies, however, have supported the concept of far worse treatment outcomes for patients with comorbid substance use The Combining Medications to Enhance Depression Outcomes (CO-MED) study reported that patients with MDD and comorbid substance use disorder were as likely to respond and remit to treatment as those with MDD alone [64], perhaps suggesting that comorbid substance abuse may be related, but not causative of treatment resistance.
Interestingly, Tamm et al. [65] reported that a childhood diagnosis of ADHD, but not cannabis use in adulthood, was associated with executive dysfunction in young adults. Thus it was suggested that earlier initiation of cannabis use (before age 16) may be linked to poor cognitive outcomes and the presence of ADHD [65].
In examining this data, it might be hypothesized that comorbid substance abuse indicative of a comorbid ADHD, and may be predictive of treatment resistant depression.
Only a few studies have investigated the moderating effects of comorbid ADHD on treatment outcomes for depression. Cunha et al. [53] reported that Cocaine Dependent Individuals (CDI) with ADHD presented with more pronounced executive alterations than and poorer cognitive functioning than controls, suggesting that pre-existing ADHD symptoms may have a significant negative impact on executive dysfunction in CDI.
Rohde et al. [66] assessed the negative impact of lifetime psychiatric comorbidity on participation in, and benefit from, CBT in adolescents with depression. Study investigators observed that patients with ADHD were more likely to relapse within the 24-month follow-up period than depressed patients without ADHD.
Birmaher et al. [67] observed response rates to paroxetine, imipramine and placebo over 8 weeks in depressed adolescents with and without ADHD. Patients with comorbid ADHD experienced significantly lower response rates, regardless of the treatment regimen, than patients with MDD alone (25% vs. 71%, respectively). Further to the point, the Treatment of Adolescent Depression Study (TADS) reported that comorbid disorders were a significant moderator of lowered response to treatment with fluoxetine, CBT, or combined fluoxetine and CBT in patients with MDD [68]. This latter study, however, did not specifically report on the moderating effects of co-morbid ADHD. It measured the moderating effect of a group of comorbid disorders that included ADHD as well as dysthymia and anxiety disorder. Taken together, the results of these studies suggest that ADHD may represent an important moderating influence on treatment outcomes for patients with MDD and comorbid ADHD. Furthermore, future studies may reveal if symptoms such as impulsivity or a pre-existing executive dysfunction could represent underlying cognitive endophenotypes that would substantially increase the risk for acquiring addictive disorders [53].
Given the high rates of comorbidity between MDD, ADHD and the suggestions that increased impairment and worse outcomes are associated with their concurrence, a great deal of research has attempted to identify the mechanisms underlying this relationship. Recent evidence from genetic, neurological, and behavioral studies has revealed altered DA-regulated reward system functioning in both MDD and ADHD [69-72]. Because DA regulates mood [8], motivation [73], attention [74], and decision making [75], alterations in DA may lead to symptoms such as negative affect, low motivation, inattention, depression, and altered hedonic responsiveness [76-80] that are common to MDD and ADHD. Recent support for this hypothesis has come from work by Silvetti et al. [81], who used the Reward Value and Prediction Model (RVPM) to examine the profile of reinforcementrelated deficits in ADHD, suggesting that anterior cingulate cortex dysregulation might play a role in the pathogenesis of motivational deficits in ADHD.
Ultimately, while the literature has traditionally suggested that the comorbidity somehow blocks the improvement of depression from happening, one might also hypothesize that the comorbidity represents a different phenotypic depression entity that will not respond as well to typical SSRIs. Understanding that phenotypic presentation would require a specific review of the features perhaps associated with the common manifestations of this presentation.
Anhedonia is generally defined as the inability to experience pleasure [82]. It is a central feature of MDD [83], but has also been studied in a variety of other illnesses including schizophrenia [84,85], substance use disorder [86,87], Parkinson’s disease [88,89], overeating [90,91], and risk-taking behaviors [92].
The term anhedonia, however, does not sufficiently capture the complex and multifaceted reward-associated deficits seen in neuropsychiatric disorders [93], but may manifest as a loss of interest or pleasure, and may preclude an individual from engaging in goal-oriented action. Investigators have identified poor hedonic responsiveness, manifested as altered sensitivity to reinforcement, in patients with ADHD [94], and suggest it as a common endophenotype for depression and the inattentive symptoms of ADHD [95].
Research into the neurobiology of anhedonia has focused on the centers in the brain related to reward and motivation, and have identified the dopaminergic and associated circuits as playing a key role [83,96,97]. Neurobiological studies have shown that lesions in reward and motivation circuits result in anhedonia [83,98-100] with neuroimaging studies demonstrating decreased activation in the medial frontal cortex as a possible causative factor underlying abnormal positive affect processing in patients [101-103].
Changes in hedonic state (ie towards or away from happiness) can be understood as positive or negative relative to their most current hedonic state, with the magnitude of the shift determining the intensity of feelings. Happiness involves a pleasant level of hedonic tone, by virtue of a shift towards the positive pole, whereas, sadness represents movement toward the opposite or negative pole. This shared hedonic dimension allows many different feelings to interact or modulate each other. Interestingly, these positive and negative shifts in hedonic tone also define reward and punishment in terms of learning. Once an association is established, it will result in the hedonic shift or outcome expected with each expression of this behavior in the future. This link between feelings and learning appear to be established by output from the limbic system, specifically the pleasure (reward) pathway [104].
One can imagine that individuals differ in their baseline range of hedonic tone, also referred to as hedonic capacity or hedonic responsiveness defined as each individual’s ability to experience pleasure [105]. Interestingly, low hedonic tone is associated with several psychopathologies in addition to MDD, including schizophrenia, and substance use [105,106]. Variations in hedonic tone are also observed in healthy subjects [107].
Perhaps then one can imagine that those born with a low hedonic capacity would be more likely to require higher volumes of positive environmental activity to raise and maintain their lower natural baseline hedonic tone to a more euthymic tone. Thus, these individuals would either endeavor to increase their lower hedonic tone through highly stimulating (likely risky behaviors) or through the use of substances such as cocaine, that heighten hedonic tone or alternatively be forced to suffer with a chronic low mood. This is seen in the poor hedonic responsiveness common to MDD and ADHD [79,94,108,109]. The hypothesis of a low hedonic tone neurobiology is centered on the belief that there is a dysfunction in the mesolimbic DA system, which of course is known to act as the central reward system [70,110]. Further support for this view has come from a variety studies that have established poor hedonic tone as a feature of both depression and ADHD [79,94,108,109,111], as well as other catecholamine dysfunctional conditions and associated prefrontal hypoactivity including schizophrenia, anxiety disorders, suicidal ideation, successful suicide, chronic fatigue syndrome, and alcohol and substance abuse [112-119]. Anhedonia is also present in Parkinson’s disease, as well as in association with dysfunctional behaviors, such as overeating, and risktaking behavior [88,90,92].
Anhedonia is a state of reduced ability to feel pleasure. It is the cornerstone symptom of depression and acts to direct research into possible treatment in treatment-resistant depression and/or double depression. Low hedonic tone, on the other hand, is the trait or genetic predisposition underlying one’s baseline range and lifelong characteristic ability to feel pleasure. Low hedonic tone would therefore be suggestive of a reduced lifetime capacity to experience euthymia and a lower likelihood of experiencing pleasure at any given time. Thus, it can be hypothesized that for those suffering with genetically lower set point or lower hedonic tone, there is an increasing likelihood of experiencing anhedonia, depression and/or dysthymia [120,121].
Thus, it follows that in response to this trait, individuals will need to do more to feel neutral or euthymic, resulting in an increased need for stimulation. In part, this may be seen as seeking external stimulation, such as through danger or risky behavior, or through internalizing (such as through fantasy), or through substance abuse (eg, cocaine), all designed to raise their current hedonic tone. Regardless of the choice to externally or internally raise their hedonic tone, patients with this trait would consistently attempt to cope by finding ways to maximize pleasure and raise mood from their low baseline tone. Furthermore, when stimulation is absent, they would experience a shift towards the more natural lower hedonic tone, and therefore a drop in their mood [122-124].
Thus much like lack of dopaminergic modulation in the dorsolateral prefrontal cortex might contribute to executive dysfunction associated with ADHD, it follows that the same deficits in dopaminergic modulation in related areas might contribute to lowered hedonic tone, increased risk of depression, treatment-resistant depression and a higher risk of substance and specifically cocaine abuse.
While the hypothesis of low hedonic tone predicting a risk of developing anhedonia, depression, or dysthymia does not provide an etiological explanation for the development of these disorders, it may provide explanation of symptom overlap between and across ADHD and cocaine use within the MDD population. Additionally, low hedonic tone may act as a predictor of the presence of ADHD within a subset of patients presenting with symptoms of MDD, dysthymia, or double depression that have been deemed resistant to traditional medication and/or CBT [95,125,126].
As well, low hedonic tone may prove to be a predictor of future vulnerabilities and therefore may be an effective target for screening that can be used in the identification of a specific subtype of depressed individuals who are likely to be treatment resistant to typical management and who may require alternative treatment choices including psychostimulants [83,95].
As such, future studies will need to be conducted in order to confirm this hypothesis in adults. In addition, monitoring children for changes in mood and/or baseline hedonic experience in relation to activity level may help illuminate specific features that can help identify and therefore build resilience or adaptive behavior before the depressive psychopathology begins. In fact, it may be possible that correct use of psychostimulants in children may not specifically treat depression on their own, but may play a role in preventing the development of depression or drug abuse comorbidities (specifically stimulants, such as cocaine and amphetamines) [127,128] as well as act as an effective adjunct to traditional psychopharmacology and/or CBT in existing patients diagnosed with depression.
As evidence emerges that ADHD in childhood and adolescence is associated with increased risk of developing MDD in adulthood [129,130], there is an urgent need to identify the pathophysiologies common to these two disorders in an attempt to understand and successfully treat them both. Converging lines of evidence identify altered dopaminergic circuits as playing a key role in the pathophysiology of these disorders, leading to low hedonic tone and anhedonia. Thus, by implementing treatment strategies aimed at correcting low hedonic tone, clinicians can achieve not only remission from ADHD but also prevent these patients from progressing to MDD later in life. For patients with MDD, correcting low hedonic tone can be seen as one strategic decision to alleviating anhedonia and may serve to treat comorbid psychopathologies.