Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2014) Volume 7, Issue 6
Leucine rich repeats (LRRs) are present in over 20,000 proteins from viruses to eukaryotes. Most LRR units are 20-30 residues long and can be divided into a highly conserved segment and a variable segment. Eight classes have been recognized. Two to sixty-two units occur in tandem to form an LRR structure. The tertiary structures of these LRRs are helical, in which the β-strands of the highly conserved segments stack in parallel. This helix consists of a super helical arrangement of repeating structural units. We call it a coil of solenoids. We have used our program HELFIT to assign helical parameters to 642 LRRs of known structures of 114 proteins. We report these parameters and their correlations with eight classes of LRR, with the number of repeat units in the LRR, with oligomerization, and with ligand state of the LRR. The helical parameters of the eight LRR classes frequently overlap one another. However, the constant distance between parallel β-strands is the primary determinant of the helical parameters of the LRRs. When the repeat number, n, in LRRs is small, the LRR structures are more variable and, by inference, more flexible. In the LRRs with n ≥ 10, Δz (the rise per repeat unit) of the “RI-like” and “Cysteine-containing” classes is smaller than those of “SDS22-like”, and “Plant-specific” classes, This difference is ascribed mainly to the difference in the structural units. The helical parameters of the LRRs unambiguously describe both right handed and left handed helices, helical dimers, and subdomains if they exist. Moreover, the helical parameters sensitively detect structural changes induced by protein, protein interactions, glycosylation, and/or mutation.
Keywords: HELFIT, LRR, Solenoid, Parallel &beta:,-sheet, BRI1, Oligomerization, Protein-protein interactions
3D: Three dimensional; AMIGO-1: Amphoterininduced Proteins 1; APL1C: Anopheles Plasmodium-responsive Leucine-rich Repeat Protein 1; AtCOI1: Coronative-insensitive Protein 1 from Arabidopsis thaliana; AtRPK2: LRR receptor-like serine/ threonine-protein Kinase RPK2 from Arabidopsis thaliana; AtTIR1: Transport Inhibitor Response 1 from Arabidopsis thaliana; AtTMK1: Probable Receptor Protein Kinase TMK1 from Arabidopsis thaliana; BAK1: BRASSINOSTEROID INSENSITIVE 1-associated Receptor Kinase 1; BRI1: Protein BRASSINOSTEROID INSENSITIVE 1; BRL1: Serine/threonine-protein Kinase BRI1-like 1; CD14: Myeloid Cell- Specific Leucine-Rich Glycoprotein; Cα: alpha-carbon; DLC1: Flagellar Outer Arm Dynein Light Chain 1; dsRNA: Double-stranded RNA; FSHR: Follicle Stimulating Hormone Receptor; FSL2: LRR Receptor- Like Serine/Threonine-Protein Kinase FLS2; Glibβ: Glycoprotein Ib beta; GPIbα: Glycoprotein Ib alpha; GPIX: Glycoprotein IX; HCS: Highly Conserved Segment; HEL: Anti-Hen Egg White Lysozyme; InlA: Internalin A; InlB: Internalin B; InlC: Internalin C; InlF: Internalin F; InlH: Internalin H; InlJ: Internalin J; InlK: Internalin K; IR: Island region; LEGL7: Legionella Leucine-Rich Repeat-Containing Protein; LGR: Leucine-Rich Repeat-Containing G Protein-Coupled Receptor; LINGO-1: Leucine-Rich Repeat and Immunoglobulin- Like Domain-Containing Nogo Receptor-Interacting Protein 1: LRIM1: Leucine-rich Immune Molecule 1; LRR: Leucine Rich Repeat; NGL-2: Netrin-G2 Ligand/ Leucine-Rich Repeat-Containing Protein 4; NGL-3: Netrin-G3 Ligand/ Leucine-Rich Repeat Containing Protein 4b; NgR-4: Reticulon-4 receptor/Nogo receptor/Nogo-66 receptor; NLRC4: NLR Family CARD Domain Containing Protein 4; NLRX1: NLR Family Member X1; PP32/ANP32A/LANP: Acidic Leucine-rich Nuclear Phospho-protein 32 Family Member A; PGIP: Polygalacturonase Inhibitor; RabGGTα: Geranylgeranyl Transferase type-2 Subunit Alpha; RanGAP: Ran GTPase Activating Protein 1; RI: Ribonuclease Inhibitor; RMSD: The Root Mean Square Deviation from the best fit helix; SERK1: Somatic Embryogenesis Receptor Kinase 1; Skp2: S-phase Kinase-Associated Protein 2; SspH: Salmonella Secreted Protein H; TAP: mRNA export factor TAP; TLR: Toll-like Receptor; Trk-A: Tyrosine Kinase Receptor A; TSHR: Thyroid-Stimulating Hormone Receptor; U2 snRNP A': U2 Small Nuclear Ribonucleoprotein A'; VLR: Variable Lymphocyte Receptor; VS: Variable Segment; YopM: Leucine-rich Effector Protein from Yersinia pestis
LRRs are present in over 20,000 proteins from viruses to eukaryotes, as listed in the data bases of PFAM, SMART, PROSITE, and InterPro [1-4]. All organisms for which sequence data are available have at least one LRR protein. Most LRR proteins are involved in protein, protein interactions, as observed in the plant immune response and the mammalian innate immune response [5-9]. Beyond immunity, extensive functional diversity occurs among LRR proteins, including their involvement in apoptosis, autophagy, ubiquitin related processes, nuclear mRNA transport, neuronal development, and the type III secretion system in pathogenic bacteria [10-16]. Furthermore, plant LRR proteins including LRR containing receptor-like kinases and LRR containing receptor like proteins act as signal amplifiers in the cases of tissue damage, of establishing symbiotic relationships, and of affecting developmental processes [17-19].
Two to sixty-two LRR units occur in tandem. Each unit is typically 20-30 residues long and can be divided into a highly conserved segment (HCS) and a variable segment (VS) [6,20,21]. The HCS consists of an eleven residue stretch, LxxLxLxxNxL, or a twelve residue stretch, LxxLxLxxCxxL, in which “L” is Leu, Ile, Val, or Phe; “N” is Asn, Thr, Ser, or Cys; and “C” is Cys, Ser or Asn. Eight classes of LRRs-“RI-like”, “Cysteine-containing”, “SDS22-like”, ”IRREKO”, “Bacterial”, “Plantspecific”, “Typical”, and “TpLRR”-have been recognized [6,20-22]. They are characterized by different lengths and consensus sequences of the VS of their repeat units. Most of the known LRRs consist of only one class of units; however a few have a mixture of two classes. A super-motif of LRRs occurs in a group of LRR proteins including the subfamily of small LRR proteoglycan including biglycan, and decorin, and TLRs 7, 8, and 9 [23-25]. Moreover, the first, N-terminal LRR unit in the “Typical” or “IRREKO” LRR domains is frequently occupied by a “Bacterial” motif [21]. Crystal and/or solution structures of representatives of all the eight classes of LRR are available.
Three residues at positions 3 to 5, xLx, in the HCS form a short β-strand [5-9]. These β-strands from tandem LRR units stack parallel and then the LRRs form an arc or a helix. This helix is not simple but consists of a super helical arrangement of repeating structural units. We call it a coil of solenoids.
The concave face, consisting of the HCSs of each LRR unit, consists of a parallel β-sheet, one strand from each HCS unit. The convex face, consisting of the VS of each LRR unit, is made of a variety of secondary structures including the α-helix, 310-helix, polyproline II helix, and an extended conformation or a tandem arrangement of β-turns (Figure 1). The various secondary structures on the convex side are connected to the strands forming the β-sheet in its concave side by two loops (Figure 1). The “ascending loop” links the C-terminal end of the β-strand in the HCS to the N-terminus of the characteristic secondary structure in the VS [9] (Figure 1). The “descending loop” links the C-terminal end of the characteristic secondary structure in the VS to the N-terminus of the β-strand in the HCS of the following unit. Most of the known LRR structures have an N- and/or C-cap that shields the hydrophobic core of the first LRR unit at the N-terminus and/or the last unit at the C-terminus [5-9]. In extracellular proteins or extracellular regions, these caps frequently consist of Cys clusters consisting of two or four Cys residues on the N- and C-terminal sides of the LRRs.

Figure 1: Diversity of secondary structures on the convex sides of LRRs in representatives of the eight classes-pig RI, Skp2, InlA, InlJ, YopM, FLS2, LINGO-1, and BACCAC_03700-for classes “RI-like”, “Cysteine-containing”, “SDS22-like”, “IRREKO”, “Bacterial”, “Plant-specific”, “Typical”, and “TpLRR” respectively. In all figures, the β-strands from two consecutive LRRs are shown. Green arrows represent β-strands, red ribbons α-helices, yellow ribbon 310-helix, pink ribbon polyproline II helix, orange tube an extended conformation, and blue tubes β-turns. Throughout the text, loops connecting the concave side to the convex side are referred to as “ascending”, and the ones connecting the convex side to the concave side are referred to as “descending”.
Non-LRR, Island Regions, IRs, interrupting LRRs are widely distributed; they are referred to as “islands” or “loop outs” [26-28]. A large number of plant LRR proteins including LRR containing receptor like kinases and LRR containing receptor like proteins have IRs [24,28]. In our analyses we treat the LRR regions on either side of its IR as distinct helices, as well as analyzing the entire LRR as a single helix.
Enkhbayar et al. [29-31] developed a program, HELFIT, that determines the helical parameters-helix axis, pitch (with handedness), radius, and number of points or units per turn-as well as RMSD. Only four points are required to define a helix. The trace of a helix with pitch=0.0Å is a circle; with N=exactly 1.0 the trace is a straight line.
Here we have determined the helical parameters for each LRR and evaluated correlation with class of LRR, with the number of repeat units in the LRR, with oligomerization, and with ligand state of the LRR to reveal some structural features of LRR structures. We demonstrate that the constant distance between parallel β-strands is the primary determinant of the helical parameters of the LRRs. When the repeat number in LRRs is small, the LRR structures are more variable and, by inference, more flexible.
A helix consisting of n repeat units may be characterized by helix axis, pitch (P), helix radius (R), and number of repeat units per turn (N). HELFIT [29-31] computes these parameters in which the helix axis is represented by the unit vector (Figure 2A). These parameters also yield the rise per repeat unit (Δz=P/N) and the rotation per repeat unit (ΔΦ=360°/N). Moreover, HELFIT gives rmsd:
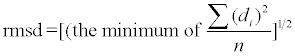

Figure 2: HELFIT analyses of LRR domains. (A) Four helix parameters directly calculated by HELFIT in BRI1 with n=25; (B) Eight representativespig RI, Skp2, InlA, InlJ, YopM, FLS2, LINGO-1, and BACCAC_03700-of each class of LRRs. Black circles indicate the first unit. In panel B, six numerical values for each protein are in order from top to bottom: the repeat number n, the helix pitch P [Å], the rise per repeat unit Δz [Å], the rotation per repeat unit ΔΦ [o], the number of repeat units per turn N [units/repeat], and the helix radius R [Å]. Left and right figures of each protein indicate side views and views along the axes of a helix, respectively. In the side views the direction of helix axis is in the direction of the arrows. In the along views the cross signs show that it is in a downward direction and also the dots show that it is in a upward direction.
where di is the closest distances from data point to the trace of the helix. Here 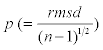 gives the regularity of helix independent of its length. If p is relatively large, the LRR is not well described by a helix and might be more easily visualized as another shape, e.g. an ellipse.
gives the regularity of helix independent of its length. If p is relatively large, the LRR is not well described by a helix and might be more easily visualized as another shape, e.g. an ellipse.
In most LRRs the three residues at positions 3-5 in the HCS form a short β-strand, which is almost completely conserved in all LRR structures [5-9]. These β-strands from adjacent repeat units form a parallel, pleated sheet on the inside, or concave, surface of the helix. As reference points for HELFIT we use the coordinates of the α-carbon (Cα) of the consensus leucine residue at position 4 (corresponding to the middle of each β-strand) in individual LRR repeat units.
Three structures of LRRs with n=3 have been determined. In these cases we performed 3D circle fitting that directly fits a circle to a set of 3D data points, because only three points define a circle [32]. The 3D circle fitting, meaning P=0.0 and Δz=0.0 as an a priori assumption determines the radius of the circle, the number per circle, the center of the circle, and the root mean square deviation from the best fit circle. The 3D circle fitting was also utilized for the estimation of the average distance between adjacent repeats by the helical parameters. Furthermore, the center of the circle was used for the calculation of the distance between the two monomers in TLR dimers.
For grouping of eight LRR classes of known LRR domains we performed similarity searches by FASTA and by BLAST using representative LRR domains clearly belonging to individual classes as query sequences. The representative LRR domains were selected from those for which the 3D structures have been solved. “RI-like” LRRs are from RI, “Cysteine-containing” from Skp2, “SDS22-like” from InlA, “IRREKO” from InlJ, “Bacterial” from YopM, “Plant-specific” from FLS2, “Typical” from LINGO-1, and “TpLRR” from BACCAC_03700.
Secondary structure assignments based on 3D coordinates were made using the DSSP program ( http://crdd.osdd.net/raghava/ccpdb/beta2_up.php) [33].
Helical parameters
Figure 2B shows HELFIT computed helices in representatives-RI, Skp2, InlA, InlJ, YopM, FLS2, LINGO-1, and BACCAC_03700-of each of the eight classes of LRRs. The fits are very good. Supplementary Table 1 shows the helical parameters–P, Δz, Δφ, N, and R of the 642 LRRs of known structures of 114 different proteins (Supplementary Table 1) [34-145].
The correlation of Δz, Δφ, and R
D is the average Cα(i)-Cα(i+1) distance between adjacent repeats, in which the Cα atoms are at position 4 in the β-strand [32]. D is a function of Δz, ΔΦ, and R.
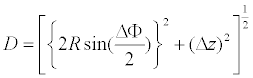 (1)
(1)
The data points of 2⋅Rsin (ΔΦ/2) versus Δz fall on a circle with radius D; although, in the “TpLRR” class the data deviate from this circle (Figure 3). The circle fittings using in total 627 data points (except for those of the “TpLRR” proteins) gives D=5.02 ± 0.00 Å [32]. This D corresponds to the inter-strand distance that is in the range of 4.5 to 5.5 Å [146]; this distance allows the formation of hydrogen bonds between parallel strands. In a canonical, parallel β-sheet the strands are aligned at the same level parallel to the axes of the individual strands. This occurs in LRR structures; the observed D is consistent with the distance between strands in the canonical parallel β-sheet.

Figure 3: The correlation of z and 2·R·sin (ΔΦ/2) in the helix parameters. (A) All 642 LRRs; (B) 139 LRRs with the repeat number n ≥ 10 in the four classes of “RI-like”, “Cysteine-containing”, SDS22-like, and “Plant-specific”. Some LRRs in the “TpLRR” class deviate from the common circle; this can be attributed to a larger twist angle of β-sheets, a breaking of β-strands in the HCS; or a prism like shape for the LRRs. “R” is the helix radius of a helix consisting of n repeat units, “Δz” the rise per repeat unit, and “ΔΦ” the rotation per repeat unit.
The D value in the “TpLRR” class is 5.52 ± 0.14 Å and thus is larger and more variable than those in other classes. The calculation of the Cα(i)-Cα(i+1) distance revealed D>6.9 Å between neighboring β-strands, as observed in FAEPRAA2165_0102, EUBVEN_01088, BACCAC_03700, and BACOVA_01565. This greater distance arises from water bridges in the hydrogen bonds between the strands.
Most of the LRRs have R=15 → 30 Å (Supplementary Table 1). However, five LRRs in human Trk-A shows very small R (=0.89 Å) [143], while LRRs 7-13 in BT_1240 has very large R (=277 Å). Even in the extreme cases the 2⋅R⋅sin (ΔΦ/2) and Δz values fall on the circle in figure 3. In the former the helix axis is nearly perpendicular to the β-strand axis. In contrast, in the latter the helix axis is nearly parallel to the β-strand axis.
The helical parameters of the eight LRR classes frequently overlap one another (Figure 2B). The Δz and 2⋅R⋅sin (ΔΦ/2) values in the “Typical” class are distributed broadly (Figure 3).
Structures of LRRs in GPIBβ, a GPIBβ/GPIX chimera, and ZP_02034617.1 with n=3 are available (Supplementary Table 1) [144]. The circle fitting analysis yields D =4.90 Å ± 0.01 Å that seems to be smaller than in LRRs with n ≥ 4. This smaller D might reflect interactions of side chains bridging N- and C-caps [144].
Left handed helices
Most LRRs form right handed helices, that is Δz>0.0 Å. However, left handed helices are adopted by LEGL7, CD14, Lmof2365_1307, FSHR, TLR1, and TLR2, in which the repeat number n ≥ 7; Δz=-0.78 → -3.17 Å (Figure 4 and supplementary Table 1).

Figure 4: HELFIT analyses of LRR domains adopting left handed helices- LEGL7, mouse CD14, Lmof2365_1307, FSHR, human TLR1, and human TLR2. Six numerical values in each protein are the repeat number and helical parameters of LRRs which in order from top to bottom the repeat number n, the helix pitch P [Å], the rise per repeat unit Δz [Å], the rotation per repeat unit ΔΦ [o], the number of repeat units per turn N [units/repeat], and the helix radius R [Å]. Left and right figures of each protein indicate side views and views along the axes of a helix, respectively. In the side views the direction of helix axis is in the direction of the arrows. In the along views the cross signs show that it is in a downward direction and also the dots show that it is in a upward direction. Black circles indicate the first unit.
Helical dimers in CD14, RI, and LGR4
In the homo-dimers of some LRRs not only the individual monomer but also the entire dimer forms a single helix. We found four examples. Mouse CD14, in which the LRR does not conform to any of the eight classes, forms a dimer in the crystallographic asymmetric unit as well as in solution [139]. Dimerization in the crystal is mediated by LRR residues in the loop between β-strands in the HCS of repeats 12 and 13. The C-terminal β-strands of the two β-sheets from the two monomers interact in an antiparallel fashion and form a large and continuous β-sheet encompassing the entire CD14 dimer. The two monomers are related by a two fold axis of rotation, perpendicular to their common helix axis. The CD14 monomer forms a left handed helix, as does the CD14 dimer (Figure 5 and supplementary Table 1).

Figure 5: HELFIT analyses of LRR domains forming helical dimers-mouse CD14, human RI, and frog LGR4. Left and right figures of each dimer indicate side views and views along the axes of a helix, respectively. In the side views the direction of helix axis is in the direction of the arrows. In the along views the cross signs show that it is in a downward direction and also the dots show that it is in a upward direction. Black circles indicate the first unit.
Human RI complexed with human angiogenin or ribonuclease I forms a dimer with human RI-angiogenin/ribonuclease I [36,37]. The whole molecule of human RI consists of only LRRs; all belong to “RIlike” class. The two complexes are held together by many hydrogen bonds formed between the N-terminal β-strands of the two human RI molecules, leading to the formation of an antiparallel β-sheet. The entire RI dimer adopts a right handed helix whose helical parameters are similar to those of its individual monomers. These two hRI molecules are related by an approximate two fold axis of rotation, nearly perpendicular to the common helical axis.
Mouse RI bound to mouse ribonuclease I forms a tetramer in the crystallographic asymmetric unit. The tetramer consists of two dimers that are related by a glide plane. The monomers of both dimers adopt a right handed helix whose parameters are quite similar to those in the dimer of human RI- angiogenin/ribonuclease I (Supplementary Table 1).
Frog LGR4, whose LRRs belong to the “RI-like” class, also forms a homo-dimer in the crystal and in solution [104]. The two monomers in the LGR4 dimer are in close proximity at their C-terminal sides and are related by a two fold axis of rotation-TLRs 1-6, TLR8, RP105, LGR4, LGR5, and Drosophila Toll, “TpLRR” class-FAEPRAA2165_01021 and EUBVEN_0 1088, and “Variable” class-BT_1240 and BAGEGG_03329. Most of the p (error) values are larger than 0.20 (Supplementary Table 1). The angle between the two helix axes (Ω) best characterizes the discontinuity (Supplementary Table 2). The LRRs with two subdomains may be grouped into four categories.
The overall shape of the entire LRR in TLRs 1-6, TLR8, RP105, and BAGEGG_03329, is well approximated as half or three quarters of an ellipse. For example, the LRRs in human TLR3 with n=25 consist of two subdomains, LRRs 1-12 and LRRs 13-25 (Figure 6). The 25 units of TLR3 forms a nearly flat ellipse with p=0.23 Å. This p value is larger than p=0.09 Å of units 1-12 in subdomain 1 and of units 13-25 in subdomain 2. Similar characteristics are observed in other LRRs that consist of two subdomains. The angle, Ω, between the two helix axes of the subdomains ranges from 13.4 → 19.2° (Supplementary Table 2). This tilt generates a larger p for the entire LRRs.

Figure 6: Schematic representations of LRRs with two subdomains-human TLR3, AtTMK1, and EUBVEN_01088. “Ω” is the angle between the two helix axes of the two subdomains.
AtTMK1, BRI1, BRL1, AtRKP2 and Drosophila Toll contain LRRs interrupted by a non-LRR, IR. For example, AtTMK1 contains 15 units interrupted by a non-LRR IR; n1=11 and n2=4 [75]. The two helix axes are nearly perpendicular to one another; Ω=74º (Figure 6 and Supplementary Table 2). The non-LRR IR has a cluster of four Cys residues with the pattern of Cx6Cx29Cx7C. The formation of a disulfide bridge in Cx7C suggests that the IR acts as an N-cap, further supporting the formation of the second subdomain. The first LRR domain in Drosophila Toll with n=19 forms a half ellipse [95]. Therefore, the LRR domain is divided into two subdomains.
EUBVEN_01088, FAEPRAA2165_01021, and BT_1240 contain LRRs in which the continuity of the parallel β-sheet is disrupted. The “TpLRR” domains in EUBVEN_01088 and FAEPRAA2165_01021 with n=14 are kinked at the central seventh and eighth repeats with 35 and 37 residues, respectively; Ω=71 → 88º (Figure 6 and supplementary Table 2). BT_1240 contains 13 LRRs of which four repeats are similar to the “TpLRR” consensus. BT_1240 forms a homo-dimer in the crystal, in which the two molecules are in close proximity at the C-terminal sides. The LRR unit 7 is 30 residues long and disrupts the continuity of the parallel β-sheet; Ω=64 → 70º (Supplementary Table 2).
The LRR domains in human LGR4 and LGR5 with n=19, and frog LGR4 with n=17 are kinked between the central eleventh and twelfth repeats [104-107]; Ω=19.1 → 40.1°. The units of LRR11 and of LRR12 do not strictly obey the LxxLxLxxNxL rule. Instead, the Asn residues are replaced by A309 and T332, respectively. As a result, the asparagine ladder (in which the Asn side chains from different turns are stacked and form hydrogen bond connecting turns) breaks in this region, thereby generating two longer β-strands [104-107].
Homo-oligomers
Proteins that contain LRRs form homo-dimers, -trimers, -tetramers, -pentamers, -hexamers, and -octamers in crystals. The LRRs of these homo-oligomers are related by non-crystallographic symmetry and hence experience slightly different packing environments. There are two patterns. The first case is that the helical parameters of individual monomers constituting the homo-oligomers are similar to each other. In this case, their repeat numbers are large, n ≥ 11. Examples include, homo-octamers-NLRC4 (n=16), Skp1 (n=11), RP105 (n=23), AtTIR1 (nn=18), and AtCOI1 (n=18); homo-hexamers-SspH1 (n=10) and biglycan (n=12); homo-pentamer-EUBVEN_01088 14; homotetramers- RanGAP1(n=11), LINGO-1 (n=14), Drosophila Toll (n=8), and CARMIL (n=16), and homo-dimers-NGL-3 (n=11) and InlA (n =16). In the second pattern the helical parameters differ significantly among the individual monomers. In this case, their repeat numbers are small; n=5 or 7. They include homo-tetramers-TAP, n=5, VLR2913, n=5, bovine coupling factor B, n=5, and human pp32, n=5; the homohexamer- human pp32, n=5; and the homo-octamer-InlK, n=7.
Eight classes of LRR
When the repeat number, n, is ≤ 8, the helical parameters for individual monomers of the homo-oligomers are sometimes variable. The helix parameters were not determined in these cases.
The consensus sequence of the “RI-like” class is LxxLxLxxNx(L/C) xxxgoxxLxxoLxxxxx. The repeat length is 28-29. Most of their VSs adopt an α-helical conformation (β-α structural units) (Figure 1). “RIlike” LRR proteins include RI (n=17), NLRC4 (n=16), CARMIL (n=16), and NLRX1 (n=8) [34-40]. The helix parameters range over: P=- 6.3 → 35.6 Å, Δz=- 0.21 → 1.76 Å, ΔΦ=11.4 → 18.5o, n=19.5 → 31.6 units/ repeat, and R=14.7 → 24.8 Å (Supplementary Table 1).
The LRR domains of RanGAP from human and fungi, n=11, and tropomodulin from chicken and Caenorhabditis elegans, n=5, show little similarity to the RI LRRs [41-45]. However, their repeat units are in β-α conformations that are the same as those of the “RI-like” class. Correspondingly, their helical parameters are similar to those of the four “RI-like” LRR proteins.
The LRR domain in LEGL7 is similar to that in CARMIL (E-value=6.2 · 10-4 in FASTA). We tentatively assign the LEGL7 LRR to the “RI-like” class. The LRR domain consists of eleven units. The consensus is VxxLxLxxNxLxxxSxxELxxxLAxIPxx with 29 residues. DSSP analysis indicates that the “ascending loops” in the eight repeats adopt a 310-helix of one turn at the underlined residues and the VS adopts an α-helix with 6 → 10 residues. The LRR domain adopts a lefthanded helix; P=-26.9 Å, Δz=-1.53 Å, ΔΦ=20.5º, n=17.6 units/repeat, and R=13.7 Å (Supplementary Table 1).
“Cysteine-containing” LRR proteins include F-box/LRR-repeat protein 20 from Rattus norvegicus, grr1 from Saccharomyces cerevisiae and grrA from Emericella nidulans. The consensus sequence is LxxLxLxxCxxITDxxoxxL(a/g)xx(C/L)xx. Tertiary structures are available for Skp2 with n=8 or 11 [46,47]. The units of Skp2 are relatively variable; they are in β-α conformations (Figure 1). The helix parameters range over: P=28.1 → 40.8 Å, Δz=1.17 → 1.75 Å, ΔΦ=15.0 → 16.2o, N=22.3 → 23.4 units/repeat, and R=16.6 → 18.9 Å (Supplementary Table 1).
The “Cysteine-containing” LRR domain in Skp2 is similar to those in AtTIR1 and AtCOI1 [48-50]. For both, n=18. The HCS of the LRR domains in AtTIR1 and in AtCOI1 are highly variable because the CxxI sequence is not conserved in most of these LRRs. In contrast, the VSs are conserved. We, therefore, assume that both the LRR domainsI1 belong to the “Cysteine-containing” class. The variable HCSs form relatively long β-strands with three to six residues and the VSs form α-helices with twelve residues. The p values are large: p=0.33 → 0.42 Å and thus the right handed helix is distorted. The helix parameters range over: P=19.3 → 20.4 Å, Δz=1.02 → 1.05 Å, ΔΦ=18.5 → 19.3o, N=18.7 → 19.1 units/repeat, and R=14.9 → 15.3 Å.
The consensus sequence of the “SDS22-like” class is LxxLxLxxNxIxxIxxLxxLxx. The repeat length is 21-23. The VSs strongly prefer a 310-helix at the underlined residues. The individual units are in β-310 conformation (Figure 1). “SDS22-like” LRR proteins include five distinct internalins (except for InlJ) with n=6 → 16 [51-68]. The “SDS22-like” LRR domains adopt a right-handed helix: P=6.27 → 73.7 Å, Δz=0.23 → 2.91 Å, ΔΦ=11.8 → 15.1°, N=23.9 → 30.7 units/repeat, and R=17.4 → 22.8 Å (Supplementary Table 1).
The LRR domain in InlK shows high similarity to that in Lmof2365_1307. These two LRR domains show significant similarity to those in InlA and internalin C2 (with E-values <10-4 in FASTA). We tentatively assume that the LRR domain in InlK and Lmof2365_1307l belong to the “SDS22-like” LRR class. Three variable HCSs at the N-terminal side adopt long β-strands with five or eleven residues. These putative “SDS22-like” domains adopt a left-handed helix; P=-62.9 → -97.4 Å, Δz=-1.90 → -3.32 Å, ΔΦ=10.9 → 14.8o, N=25.8 → 33.1 units/ repeat, and R=12.9 → 24.7 Å.
The consensus sequence of the “IRREKO” class is LxxLxLxxNxLxxLDLxx(N/L/Q/x)xx or LxxLxCxxNxLxxLDLxx(N/ L/x)xx [21]. The only available 3D structure is that of InlJ [69]. InlJ with n=15 belongs to this class; although, the first and the last units are “SDS22-like”. The VS of TxLDL(T/S)x(N/L)Tx adopt an extended β conformation and β-turns (Figure 1). Its helical parameters are P=73.3 Å, Δz=2.80 Å, ΔΦ=13.7º, N=26.2 units/repeat, and R=17.7 Å (Supplementary Table 1).
The consensus sequence of the “Bacterial” class is LxxLxVxxNxLxxLP(D/E)LPxx. The repeat length is 20-22. The structural units are in β-polyproline II helix conformation (Figure 1). “Bacterial” LRR proteins include YopM (n=16), SspH2 (n=13), SspH1 (n=10), and ipaH3 (n=9) [147-150]. The helix parameters range over: P=46.7 → 103 Å, z=1.64 → 2.61 Å, ΔΦ=8.71 → 12.7°, N=28.4 → 36.0 units/repeat, and R=20.6 → 24.6 Å (Supplementary Table 1).
“Plant-specific” LRR proteins include protein FLS2, BRI1, BRL1, BAK1, SERK1, AtTMK1, AtRPK2, and PGIP [70-77]. BRI1 and BRL1 are homologs. BAK1 and SERK1 are also homologs. The consensus of the “plant-specific” class is xxLxLxxNxLxGxLPxxLxxLxx with 24 residues [22,151]. The AtTMK LRR domain with n=15 is kinked at the central eleventh and twelfth repeats [75]. The consensus sequence, LxGxLP, at positions 11 to 16 forms a second parallel β-strand in the “ascending loop”. Thus, the structural unit is β-β-310 (Figure 1). The helix parameters with n=9 → 29 range over: P=57.2 → 87.0 Å, Δz=2.39 → 3.37 Å, ΔΦ=13.7 → 15.0°, N=24.0 → 26.2 units/repeat, and R=15.2 → 17.5 Å (Supplementary Table 1).
“Typical” LRRs are the most abundant LRR class [6,20]. The consensus sequence is LxxLxLxxNxLxxLpxxoFxxLxx in which uppercase indicates more than 50% occurrence of a given residue in a certain position; lowercase indicates 30-50% occurrence; “L” is Leu, “N” is Asn, “p” is Pro, “o” indicates a non-polar residue, “F” are Phe, and “x” is a non-conserved residue. “Typical” LRR proteins include 53 proteins (Supplementary Table 1) [71, 78-137].
Most of their VSs adopt a tandem arrangement of three β-turns (which we call consecutive -turns) on the convex faces (Figure 1). The consecutive -turns form a flattish amphipathic structure with main chain hydrogen bonds in a linear arrangement. Adjacent β-turn repeat motifs form a regular parallel packing arrangement with the core hydrophobic residues pointing alternately in and out of the plane of the structure interlocking with the corresponding hydrophobic residues from the adjacent repeat. The consecutive -turns have already been reported in the underlined eight residues of the VS consensus of xxLPxGLLxGLxx seen in three LRR units in GPIbα with n=9 [125]. Thus, the structural units may be represented by consecutive -turns.
Phenylalanine at position 19 is internally buried and is surrounded by four Leu’s, which form the hydrophobic core of the LRR structure. These Phe’s of consecutive units form the spine of the LRR [89]. This Phe spine involves aromatic, aromatic interactions; the benzyl groups form stacks of Phe side chains. The first, N-terminal LRR unit in most of the “Typical” LRR domains is “Bacterial” LRR. The VS adopts the polyproline II helix instead of consecutive β-turns.
The helix parameters with n=7 → 27 range broadly: P=- 62.0 → 93.2 Å, Δz=- 1.53 → 3.24 Å, ΔΦ=7.41 → 15.0o, N=23.9 → 51.2 units/repeat, and R=15.5 → 38.9 Å (Supplementary Table 1). The LRR domains may be grouped into a right handed helix, a left-handed helix, and a near flat.
Human Slit2 contains four tandem arrays (D1 to D4) of LRRs that consist of six to eight units. The structures of D2, n=7, D3, n=7, and D4, n=6, have been determined in the free state [91-93]. The helical parameters differ significantly from each other; D2>D4>D3 in P; D4>D2>D3 in both Δz and ΔΦ, and D3>D2>D4 in both N and R (Supplementary Table 1).
The LRR domains in biglycan, decorin, and TLR8 consist of three or four tandem repeats of a super motif of STT in which “S” is a “Bacterial” LRR unit and “T” is a “Typical” LRR unit [23,24]. The LRR domains of biglycan and decorin with n=12 consist of four repeats of STT. Also the N-terminal LRR domain in TLR8, n=27, contains three repeats of STT. The helix parameters range over: P=36.7 → 51.8 Å, Δz=0.82 → 1.84 Å, ΔΦ=10.5 → 14.3°, N=25.3 → 34.3 units/repeat, and R=19.0 → 26.3 Å.
The LRR domain in Xcv3220, n=9, has high sequence similarity to those in both human LGR4 with E-value=7.7 · 10-6 in FASTA and human LGR5 with E-value=7.0 · 10-6 in BLAST. We assume that this Xcv3220 LRR domain belongs to the “Typical” class. Seven of the nine repeats are represented by the consensus of LxxLxLxxCxx(x/-) LxxLPxxLxxLxx with 23-24 residues. The structural unit is β-310. The LRR domain adopts a left-handed helix; P=22.3 Å, z=0.59 Å, ΔΦ=-9.6º, N=37.6 units/repeat, and R=29.7 Å.
The consensus sequence of the “TpLRR” class is LxxLxLxxxLxxIgxxAFxx(C/N)xx. The repeat length is 23-25. Bacterial “TpLRR” proteins include BACOVA_04585, bacterial group 3 Ig-like proteins (BACCAC_03700 and BACOVA_01565), FAEPRAA2165_01021, EUBVEN_01088, and BT_1240. The repeat numbers range from 12 to 14. The tertiary structures of the six “TpLRR” proteins have been determined. The ascending loops consist mainly of one β-turn and short β-strand of two residues, while the most of the descending loops consist of a single β-turn (Figure 1). Most of their VSs adopt a tandem arrangement of two or three β-turns.
The helix parameters are variable among the six proteins and deviate significantly from those of the other classes (Supplementary Table 1). This can be attributed to a larger twist angle of β-sheets, to a breaking of β-strands in the HCS, or to a prism like shape for the LRRs. The mean twist angle of β-sheets in LRR proteins belonging to the other classes was 3 → 8° [32]. LRRs in the “TpLRR” class sometimes do not form β-strands in the HCS.
Most of LRRs in BT_0210, n=18, BAGEGG_03329, n=18, and LRIM1, n=11, are highly variable and could be assigned to none of the eight classes [134,138-144]. Their helix parameters range over: P=66.2 → 96.2 Å, Δz=1.66 → 2.88 Å and ΔΦ=8.5 →10.8°, N=33.4 → 42.1 units/ repeat, and R=22.0 → 31.8 Å (Supplementary Table 1). These parameters are most similar to those in the “Bacterial” domains.
Structural change induced by protein, protein interactions, glycosylation and/or mutation
The LRRs of VLR and Slit2 belong to the “Typical” class, as noted. VLRB.2D, n=5, bound to a protein antigen, HEL, as well as nonbound VLR has been determined [83]. VLR uses nearly its entire concave surface to bind HEL. This binding causes large changes of the helical parameters; a decrease of P, Δz, and ΔΦ, and an increase in R (Supplementary Table 3). The structures of D3 in human Slit2 have been determined in the free state and in the complex with the Ig domain from Robo1 [91]. As seen in the VLRB.2D-HEL complex, the Slit2 D3 domain uses its concave surface to bind Ig. The formation of the complex causes large changes of the helical parameters. Conversely, an increase of P, Δz, and ΔΦ, and a decrease in R is observed (Supplementary Table 3). The same structural change occurs in the InlA-cadherin complex; although, it is minor (Supplementary Tables 1 and 3) [56].
The crystal structures of free LRIM1 and of free APL1 and of their hetero-dimer complex have been determined [134]. The LRIM1/ APL1complex has a single intermolecular disulfide bond. LRIM1 and APL1 contain 11 and 15 repeat units, respectively. The HELFIT analysis detects structural change in both LRR domains due to the complex formation. The LRIM1 LRR domain in the complex shows a significant increase in P and Δz and decrease in R (Supplementary Table 3). In contrast, the helical parameters in the APL1 LRR domain are reversed and have small changes in Δz and R.
The structures of frog LGR4 have been determined in the free state and in complex with R-spondin [104]. The formation of the complex induces small structural changes and increases in P, Δz, and R (Supplementary Table 3).
The structures of free RabGGTα and its complex bound to escort protein 1 have been determined [53,54]. The helical handedness of the LRR domain with n=6 reverses upon binding to REP (Supplementary Table 1).
The crystal structures of the complexes of Skp1-Skp2 and Skp1- Skp2-Cks1 are available [46,47]. The binding of Cks1 causes slight structural changes; an increase of P, Δz, and ΔΦ, and a decrease in R. Moreover, the crystal structures of the complexes with Ran-GPPNHPRanBP1- RanGAP and Ran-GDP-AlFx-RanBP1-RanGAP have been determined [43]. The helical parameters of these two complexes differ slightly from one another.
Many structures of GPIbα have been reported in the free state and in complexes with one or two α-thrombins, von Willebrand Factor A1, and PEP inhibitor [124-131]. We do not find a clear pattern of structural changes. However, it appears that the binding of two α-thrombins causes a structural change.
The crystal structures of NgR-4, n=10, in both the non-glycosylated state and N-glycosylated at Asn-82 and Asn-179, are available [88,89]. N-glycosylation induces a decrease of P, Δz, and ΔΦ and an increase of N and of R (Supplementary Table 3). The G28E mutant of bovine coupling factor B, n=5, has a smaller R than does the wild type (Supplementary Table 1) [152].
Spatial arrangement of TLR dimers
The structures of the unliganded and ligand activated human TLR8 dimer were determined [119]. Helix parameters of the two monomers in the unliganded dimer are generally similar to those in the liganded dimer. However, upon ligand binding the spatial arrangement of the two monomers in the dimer changes. The distance between the two monomers at their C-terminal regions changes from 53 Å to 30 Å [119]. HELFIT calculates the distance and the angle between the two helical axes of the two monomers in the dimers, L and ψ. Correspondingly, the ligand binding drastically changes these two parameters; ΔL=- 2.6 Å and Δψ=17º (Figure 7).

Figure 7: Schematic representation of spatial arrangement of unliganded and ligand activated human TLR8 dimer. “L” and “ψ” are the distance and the angle between the two helical axes of the two monomers in the dimers, respectively. Left and right figure indicate the unliganded and the liganded TLR8 dimer.
The structures of TLR3 complexed with dsRNA and with six different Fabs (Fab15 light and heavy chains, Fab12 light and heavy chains, and Fab1068 light and heavy chains) have been determined, as well as the structure of free TLR3 [112,113]. The formations of the complexes induce decreases in the helix radii. The dsRNA TLR3 dimer has L=74.5 Å and ψ=148º.
Furthermore, the hetero-dimers of human TLR1 and TLR2, and of mouse TLR2 and TLR6 have been determined [108,109]. The values of L and ψ are 44.0 Å and 168º in the TLR1-TLR2 dimer and 53.0 Å and 169º in the TLR2-TLR6 dimer, respectively.
The constant inter-strand distance is the primary determinant of the helical parameters of the LRRs
The HELFIT analysis demonstrates that the inter-strand distance (D) is constant in the seven classes except for the “TpLRR“ class; D=5.02 ± 0.00 Å (Figure 2B). In contrast, D in the “TpLRR” class is significantly larger; D=5.52 ± 0.14 Å. This can be attributed to non-uniformity of the inter-strand distance. The helix radius R in some LRRs belonging to the “TpLRR” class is extremely small or very large (Supplementary Table 1). Thus, the overall shape of these LRRs of the “TpLRR” class is best described as a prism; while those of the other classes are helices or semi-ellipses.
The “ascending loops” influence the helical parameters of the LRRs
The LRRs in RI and LEGL7 belong to the “RI-like” class. The RI LRR adopts a right handed helix or a horse shoe shape. In contrast, the LEGL7 LRR adopts a left handed helix (Figure 4). The LRRs form β-strands on the concave face (in the HCS) and α-helix of 9 → 15 residues on the convex face (in the VS). Both of the “descending loops” also consist of one β-turn. On the other hand, the ascending loops of the LEGL7 LRR include a 310-helix of one turn; while the RI LRR does not adopt this helix in the loops.
Some LRRs in NLRC4, InlK, Lmof2365_1307, AtTIR1, AtCOI1, TLRs 1-6, and TLR8 do not obey the LxxLxLxxNxL consensus, especially at the underlined residues. In addition, some LRRs in these proteins frequently form longer β-strands consisting of four to seven residues in the HCS. This affects not only the ascending loops but also the descending loops. Indeed, the right handed helices of NLRC4, AtTIR1, AtCOI1, TLRs 1-6 and TLR8 are distorted. Also the LRRs in InlK and Lmof2365_1307, which belong to the “SDS22-like” class, adopt left handed helices; while those in the normal “SDS22-like” class form right handed helices.
The HCS of the “TpLRR” class with ten residues is shorter than eleven or twelve residues in the “Typical” class. On the other hand, the VS of the “TpLRR” class is quite similar to that of the “Typical” class. Both VSs consist mainly of consecutive -turns (Figure 1). However, the helical parameters indicate that the “Typical” class adopts a helix, while the “TpLRR” class resembles a prism. These observations can be attributed to a structural difference of the ascending loops and the inter-strand distance.
The helix parameters vary among the “Typical” LRRs. Seven of the ten repeats in NgR-4 and eleven of the fourteen repeats LINGO-1 conform to the “Typical” consensus. However, their structures differ from one another. The NgR-4 LRR domain adopts a right handed helix with Δz=2.09 Å; while the LINGO-1 LRR arc is nearly flat; |Δz|=0.06 Å. The LINGO-1 LRRs show a structural periodicity that consists of four tandem repeats of three consecutive LRR units. The first and second VSs in the three LRR units adopt a β-strand plus consecutive β-turns, while in the last VS the corresponding β-strand breaks. In contrast, this periodicity is not seen in the corresponding residues in NgR-4. The different helical parameters may be due to a difference in structure of the ascending loops and the conformations on the convex face.
In conclusion, the above observations suggest that the helical parameters more strongly depend on the structures of the “ascending loops” than of the “descending loops”. Moreover, the helical parameters of the LRRs are influenced by helical elements on the convex face and the uniformity of parallel strand stacking on the concave face.
LRR domains having small repeat number are highly flexible
The values R and Δz versus n for all 642 LRRs show a lot of variation, if n is small (Figure 3). Here we describe some examples. The first is the family of VLR. The lysozyme bound VLRB.2D forms a tetramer in the crystal [83]. The two monomers adopt a right handed helix, while the other two monomers adopt a left handed helix. Sea lamprey VLRB, n=5, bound to the BclA protein forms a trimer [84]. One monomer forms a right handed and the other monomer forms a left handed helix. The third monomer is not visible in the electron density map of the crystal structure. Tetrameric VLR 2913 has a range of helical parameters in its four monomers (Supplementary Table 1).
The crystal structures of InlB, n=8, in wild type, mutant, and complexes have been determined [60-64,85]. The InlB molecules form a monomer, dimer, and hexamer in the crystals. The helical parameters show a range of variation. InlB with an additional LRR unit inserted, n=9, forms a trimer. All the monomers have similar helical parameters.
Right handed and left handed helices exist in the dimer of SERK1, n=5, bound to BRI1 [71] and in the tetramer of bovine Coupling Factor B, n=5 [152]. InlK, n=7, forms a homo-octamer in the crystal [68]. The helix parameters of these eight monomers are highly variable (Supplementary Table 1). Even with n=17 in RI, the helix parameters are significantly different among four vertebrate species: both P and Δz decrease in order of pig, bovine, human, and mouse [34-37].
These observations strongly suggest that LRR domains with n ≤ 7 are more variable and are more flexible in solution. NMR data of LC1 with n=6 support this inference; within the β-sheets the consensus leucine residues at position 4 have relatively low order parameters [52].
Structural features of individual LRR classes
As noted, the helical parameters of the eight LRR classes frequently overlap one another. Moreover, when the repeat number in LRRs is small, the LRR structures are more variable. Taken together, the helical parameters of the LRRs with n ≥ 10 were compared to find the structural features (Figure 3B and supplementary Figure 1). The “TpLRR” class deviates significantly from other classes. The “Typical” class has highly variable values. The rise per subunit, Δz, of the “RI-like” and “Cysteinecontaining” classes is clearly smaller than those of the “SDS22-like”, and “Plant-specific” classes. This is mainly ascribed to the difference in the structural units.
Structures of non-LRR, IRs
The structures of non-LRR, IRs may be considered in two groups. In the first group, IRs allow the continuity of the parallel β-sheet and the LRR units to form a single domain of a regular LRR structure. Examples include the IRs of BRI1, AtRPK2, BT_0210, and TLR8.
The BRI1 LRR domain has 25 units interrupted by a 70 residue IR between unit 21 and unit 22 [28]. B. thetaiotaomicron BT_0210 contains 18 units interrupted by a 76-residue IR between units 2 and 3. In the two LRR domains the helical handedness reverses (Supplementary Table 1). The IR forms a small domain those folds back into the interior of the helix, where it has extensive polar and hydrophobic interactions. The IR between repeat units 15 and 16 of TLR8 is 39 residues long [24]. The additional residues loop out from the expected helical path of a regular LRR before rejoining it some residues later [119]. Consequently, this IR is in the exterior of the helix, in contrast to the IRs in BR11 and BT_0210.
In the second case IRs disrupt the continuity of the parallel β-sheet and form two distinct subdomains. This case is observed in AtTMK1 [75] and Drosophila Toll [95]. The AtTMK1 LRR domain contains 15 repeats interrupted by a 41 residue IR between LRR11 and LRR12 [28], while the Toll LRR domain with 24 repeats has a 68 residue IR between LRR10 and LRR11[95].
We have assigned helical parameters to the 642 LRRs of known structure using the program HELFIT. These parameters and their correlations with class of LRR, with the number of repeat units in the LRR, and with oligomerization and ligand state of the LRR are described. The helical parameters of the eight LRR classes frequently overlap one another. However, the constant distance between parallel β-strands is the primary determinant of the helical parameters of the LRRs. When the repeat number, n, is small, the LRR structures are more variable and, by inference, more flexible. In the LRRs with n ≥ 10, rise per repeat unit, Δz, of the “RI-like” and “Cysteine-containing” classes are smaller than those of “SDS22-like”, and “Plant-specific” classes. This is ascribed to the difference in the structural units. The helical parameters of the LRRs unambiguously define both right handed and left handed helices, and helical dimers and subdomains if they exist. Moreover, the helical parameters sensitively detect structural changes induced by protein, protein interactions, glycosylation, and/or mutation.
We are grateful to Tuvdendorj Nomin-Erdene for her help of the HELFIT analysis. We also thank Chai JiJie of Tsinghua University for his providing of the PDB file of AtRPK2.