Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2009) Volume 2, Issue 1
As drug resistance problem persists in case of Leishmaniasis, modeling and analysis of different essential proteins of Leishmania strains will help us further to discover novel lead compounds. Lipophosphoglycan 2 (LPG2) protein is required for the development of Leishmania throughout their life cycle, including for virulence to the mammalian host. LPG2 participates in a specialized virulence pathway, which may offer an attractive target for chemotherapy. Homology models of LPG2 of five Leishmania species have been constructed using the X-ray structures of different transporter proteins as templates, by comparative protein modeling principles. The resulting model has the correct stereochemistry as gauged from the Ramachandran plot and good three-dimensional (3-D) structure compatibility as assessed by the Procheck and Profiles-3D scores. Functional assignment of LPG2 protein of Leishmania strains by SVM revealed that along with transporters activity it also performs several novel functions e.g. iron-binding, sodium-binding, copper binding. It also belongs to protein of major facilitator family (MFS) and type II (general) secretory pathway (IISP) family. Important functional motifs have been identified in LPG2 protein of different Leishmania strains using different programs. Potential Ligand Binding Sites (LBSs) in LPG2 protein of these strains have been identified using Pocket Finder program. On the basis of structure of ligand binding sites, particular LPG2 inhibitors can be designed. The similarity in the molecular structure, function and differences in LBSs of LPG2 of L. donovani, L. major, L. infantum, L. braziliensis and L. mexicana provide evidences for selective and specific LPG2 inhibitors.
Keywords: LPG2 protein, Leishmaniasis, Comparative (homology) modeling, Phyre (Protein Homology/analogY Recognition Engine), SVM (Support Vector Machine), Ligand Binding Sites (LBSs).
Leishmaniasis is identified by clinical syndromes caused by obligate intracellular protozoa of the genus Leishmania and transmitted from one host to another by the bite of blood sucking sand fly vectors. Visceral leishmaniasis also known as Kala-Azar (KA) is caused by Leishmania donovani and is fatal if it remains untreated (Bhattacharyya et al., 2002). It is typically a vector-borne zoonosis, with rodents as common reservoir hosts and humans as secondary hosts. Visceral leishmaniasis (VL), the most severe form (which is usually fatal if patients are untreated), which is due to Leishmania donovani, is common in less developed countries (Paris et al., 2004). Leishmania is endemic in large parts of the world with 600,000 new clinical cases reported annually and possibly more unreported (Vergnes et al., 2007).
A short list of drugs includes SAG (Sodium Antimony Gluconate), amphotericin-B, pentamidine, and the oral drug miltefosine, which is in phase IV clinical trial in India (Bihar, Patna). Already a decrease in efficacy has been noted against this novel molecule (Croft et al., 2006). A comparative analysis of a genetically related pair of Sb (V)-sensitive and -resistant Leishmania donovani strains isolated from kala-azar patients revealed that the resistant isolate exhibited cross-resistance to other unrelated Leishmania drugs including miltefosine and amphotericin-B (Vergnes et al., 2007).
Lipophosphoglycan (LPG) is the major cell surface molecule of promastigotes of all Leishmania species. It is comprised of three domains i.e. a conserved GPI anchor linked to a repeating phosphorylated disaccharide (P2; PO4-6-Gal (β1-4) Man (α1- ) backbone variously substituted with galactose, glucose and arabinose residues in L. major and capped with a neutral oligosaccharide (Ng et al., 1994). The main surface glycoconjugate on promastigotes, lipophosphoglycan (LPG), is crucial for parasite survival (Winberg et al., 2007).
LPG2 encodes a 37 KDa protein of 341 amino acids, containing up to 10 transmembrane domains (Descoteaux et al., 1995; Ma et al., 2004). LPG2 is a member of a growing family of genes implicated in nucleotide-sugar transport. The family is large, covers several nucleotide-sugar specificities and is evolutionarily diverse including Leishmania, yeast,C. elegans, plants and humans (Ma et al., 2004). Because of its hydrophobicity, subcellular location, and similarity to other proteins implicated in transmembrane transport, LPG2 protein is golgi GDP-mannose transporter required for addition of disaccharide-phosphate units on lipophosphoglycan and related glycoconjugates (Descoteaux et al., 1995; Ma et al., 2004). The amino acid sequence of SQV-7 protein of C. elegans and Leishmania donovani protein, LPG2 are similar to each other (67 (20%) are identical), which is required for transport of GDP-mannose across membranes (Descoteaux et al., 1995; Ma et al., 2004). Such transporters are required to bring nucleotide sugars from the cytosol, where they are synthesized, into the endoplasmic reticulum and Golgi apparatus, where they are used as sugar-donor substrates by glycosyltransferases (Abeijon et al., 1997). The hydropathy plots of SQV-7 and LPG2 are highly similar (Kyte et al., 1982). Human cells have no detectable GDPmannose transport activity, yet there are at least two human proteins similar to LPG2; thus, it is likely that LPG2, SQV-7, and the two human proteins are members of a family of transporters that have a variety of nucleotide-sugar specificities (Ma et al., 2004).
For virulence and transmission, the protozoan parasite Leishmania assembles a complex glycolipid on the cell surface, the lipophosphoglycan (LPG). Functional complementation identified the gene LPG2, which encodes an integral golgi membrane protein implicated in intracellular compartmentalization of LPG biosynthesis. Ipg2- mutants lack only characteristic disaccharide-phosphate repeats, normally present on both LPG and other surface or secreted molecules
considered critical for infectivity. In contrast, a related yeast gene, VAN2/VRG4, is essential and required for general golgi function. These results suggest that LPG2 participates in a specialized virulence pathway, which may offer an attractive target for chemotherapy (Descoteaux et al., 1995).
Epitope tagging experiments localized the LPG2 protein to the parasite’s golgi apparatus, with the C-terminus located on the lumenal side (Descoteaux et al., 1995). Transient transfection of LPG2 expression constructs, suggested that LPG2 acts autonomously as the GDP-Man transporter. It is reported that LPG2 occurs in a hexameric complex inLeishmania and also showed that GDP-Man, GDP-Ara, and GDP-Fuc can be transported by this NST. These findings have important implications to the structure and function of the NST family in both Leishmania and other eukaryotes (Hong et al., 2000).
Entry of Leishmania into visceral organs can cause damage to visceral organs and neurodegenerative symptoms are likely to occur. Hence there is requirement of study on structural and functional characteristics of different proteins of Leishmania strains to target the protein to find a suitable anti-leishmanial drug. X-ray crystallographic structure is not available for this important protein of Leishmania species. Modeling of the LPG2 protein, assigning function to this protein, identifying different ligand binding sites will give us useful information regarding LPG2 protein.
Structural Modeling
The sequence of LPG2 protein (341 amino acids) of Leishmania donovani was downloaded for structural modeling from NCBI. Multiple alignments of the related sequences were performed using the online available ClustalW program accessible through the European Bioinformatics Institute (Thompson et al., 1994; http://www.ebi.ac.uk/Tools/clustalw2/index.html). No X-ray crystallographic or NMR structure of this protein of any Leishmania species has yet been determined. Tertiary structures of LPG2 protein of different Leishmania strains were modeled on the basis of different template structures from PHYRE, I-Tasser. Structure validation was performed using ANOLEA, Profiles- 3D, WHATIF, and Model-3D, molecular modeling tool (Profiles- 3D) of discovery studio.
Transmembrane Region Prediction
Different servers i.e. TMHMM, SOSUI, HMMTOP and TMpred servers were accessed to validate the TM region of LPG2 protein (Krogh et al., 2001; Hirokawa et al., 1998;TusnaÂdy et al., 1998; Hofmann et al., 1993). TMHMM, a new membrane protein topology prediction method, is based on a hidden Markov model.
Ligand Binding Site Prediction
Pocket-Finder is a pocket detection algorithm based on Ligsite written by Hendlich et al (1997). Pocket-Finder works by scanning a probe radius 1.6A° along all gridlines of grid resolution 0.9 A° surrounding the protein. The probe also scans cubic diagonals. Grid points are defined to be part of a site when the probe is within range of protein atoms followed by free space followed by protein atoms. Grid points are only retained if they are defined to be part of a site at least five times (Hendlich et al., 1997).
Protein Function Assignment of LPG2 Protein by SVM
To know novel functions of LPG2 protein of different Leishmania strains were searched at BIDD (Cai et al., 2003; http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi). The web-based software, SVMProt, support vector machine (SVM) classifies a protein into functional families from its primary sequence based on physico-chemical properties of amino acids (Cai et al., 2003). Novel protein function assignment of different proteins of SARS virus and Japaneseencephalitis virus has already been reported (Cai et al., 2005; Sahoo et al., 2008).
ELM (Eukaryotic Linear Motif) Server
Functional sites in eukaryotic proteins which fit to the description “linear motif” are currently specified as patterns using regular expression rules. ELM server provides core functionality including filtering by cell compartment, phylogeny, globular domain clash (using the SMART/ Pfam databases) and structure (Puntervoll et al., 2003). Individual functions assigned to different sequence segments combine to create a complex function for the whole protein.
PredictProtein Server
Predict Protein provides PROSITE sequence motifs, lowcomplexity regions (SEG), nuclear localization signals, regions lacking regular structure (NORS) and predictions of secondary structure, solvent accessibility, globular regions, transmembrane helices, coiled-coil regions, structural switch regions, disulfide-bonds, sub-cellular localization, and functional annotations (Puntervoll et al., 2003; Rost et al., 2004; Bairoch et al., 1997; Ceroni et al., 2004).
Structure, function and ligand binding site analysis of LPG2 protein will lead to identification of novel targets for design of suitable lead compounds inhibiting the specific functions of L. donovani, L. major and L. infantum.
Transmembrane Region Prediction
Different servers have been accessed for accurate prediction analysis of transmembrane region e.g. TMHMM, HMMTOP, SOSUI and TMpred. Trans-membrane prediction analysis found that LPG2 protein of L. infantum and L. donovani are having same number of transmembrane regions (ten) and involvement of particular amino acids in the TM regions is similar. The LPG2 sequences of L. infantum and L. donovani are very similar to each other except at two positions 220 and 221 where threonine (T) is replaced by isoleucine (I) and methionine (M) respectively in L. infantum. Even if the number of transmembrane region are same (ten) in L. mexicana, L. donovani and L. infanum, it is found that 3rd and 4th transmembrane regions are different (3rd TM region comprises of aa 72-94 and 4th TM comprises 98-120 amino acids) in L. mexicana whereas in L. infantum and L. donovani it is coded by 77-99 and 101- 123 amino acids. LPG2 of L. braziliensis and L. major both comprise of nine TM regions but the transmembrane regions are quite different, only in few cases these are similar. Involvement of different amino acids in formation of nine different trans-membrane regions of different Leishmania strains is shown in (Fig.1).

Figure 1: Involvement of different amino acids in formation of nine trans-membrane regions of LPG2 protein of different Leishmania strains.
Multiple alignment of amino acid sequences of LPG2 protein of different Leishmania strains shows that they are very close to each other ranging from 78-99% (Table 1). Multiple alignment of LPG2 protein shows that Ldv and L. infantum are having 99% identity (Fig. 2). From phylogram, LPG2 protein of L. braziliensis is found to be far from other Leishmania strains (Fig.3). Many amino acid changes are found to be present towards the carboxy terminus in the annotated amino acid sequences.
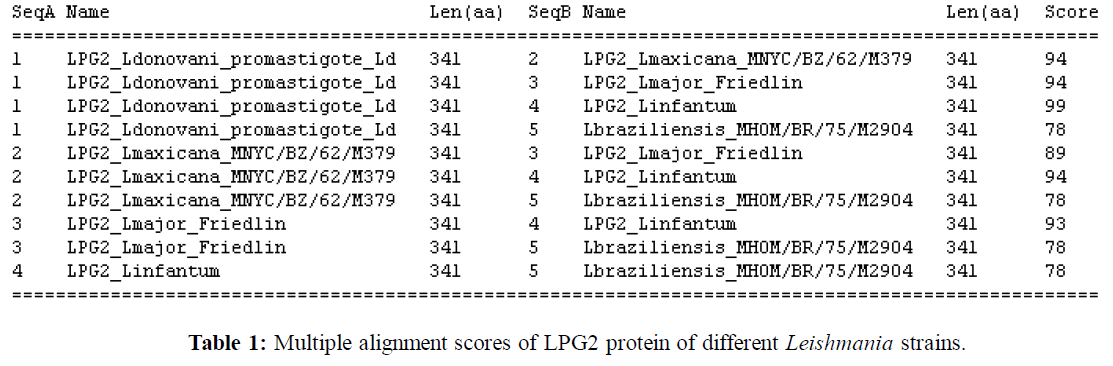

Figure 2: Multiple alignment of amino acid sequences of LPG2 protein of different Leishmania strains.

Figure 3: Phylogram showing phylogenetic relationship of LPG2 protein of different Leishmania strains (L. donovani, L. major, L. infantum, L. mexicana and L. braziliensis).
Structure Analysis of LPG2 Protein
Five different models of LPG2 protein of different Leishmania strains were screened for profiles-3d score of DS (Accelrys). The best model of different Leishmania strains were screened by profiles-3d score and the best was selected for further analysis. About 14 - 27 helices have been predicted for LPG2 protein of different models of variousLeishmania strains. The best model of LPG2 protein of L.infantum contains 22 helices, whereas 19 helices are there in case of L. donovani and L. major. The models for LPG2 protein of different Leishmania strains are shown in Fig 4. The profiles-3D scores of best predicted models of LPG2 protein of various Leishmania strains (Table 2), shows that highest score (111.29) has been found in case of LPG2 protein model of L. major and lowest score (91.23) has been found in case of L. braziliensis. Invalid regions have been detected in different models. Highest numbers of invalid regions have been found in one model of L. mexicana whereas lowest number of invalid regions is found in other models of L. mexicana, L. major and L. donovani. On further side chain and loop modeling refinements of the predicted models of LPG2 protein, the profiles-3D score is found to be decreased. In Ramachandran plots (Procheck), 88- 91% residues belong to core region, 6-8 % residue in allowed region, 1-2.5% in generously allowed regions and 1- 3% in disallowed regions (Table 3). From Ramachandran plot, it is known that maximum residues in LPG2 protein are responsible for formation of helices. Also transmembrane region prediction analysis detected nine or ten transmembrane regions to be present in LPG2 protein of all these strains. Hence there is involvement of helices in formation of LPG2 protein.

Figure 4: Ribbon representations of the modeled LPG2 protein images of all different Leishmania strains using Discovery Studio 2.0 (Accelrys) software (a) L. donovani, (b) L. major, (c) L. infantum (d) L. mexicana and (e) L. braziliensis.
| Model Features Strain Names | Number and percentage of alpha helices |
Number and percentage of 3,10(310) helices | Number of chains | Profile 3-D scores |
| L. major | 17 / 68.9% 7(min)-27(max) residues take part in formation of helices |
2 / 1.8% 3 residues |
1 | 111.29/2 (Model 5) |
| L. mexicana | 17/ 73% 5(min)-28(max) residues take part in formation of helices |
1 / 0.9% 3 residues |
1 | 101.8/2 (Model 5) |
| L. infantum | 21/ 70.4% 4(min)- 23(max) residues take part in formation of helices |
1 / 0.9 % 4 residues |
1 | 108.48/8 (Model 4) |
| L. braziliensis | 19/ 69.2% 4(min)- 24(max) residues take part in formation of helices |
2/ 1.5% 3 residues |
1 | 91.23/13 (Model 3) |
| L. donovani | 24 / 68.3 % 5(min)- 16(max) residues take part in formation of helices |
3 / 2.3% 3-4 residues |
1 | 101.8/2 (Model 5) |
Table 2: Promotif search result summary and Profiles-3D scores of modeled structures of LPG2 proteins of different Leishmania strains.
| Residues | Number of Amino acids involved Lbrzl Lmjr Linf Lmx Ldv |
Percentage of amino acids involved Lbrz Lmjr Linf Lmx Ldv |
| Residues in most favoured regions [A, B, L] | 270 272 271 276 263 | 88.2 88.6 88.6 91.1 86.8 |
| Residues in additional allowed regions[a,b,l,p] | 28 25 20 19 28 | 9.2 8.1 6.5 6.3 9.2 |
| Residues in generously allowed regions [~a,~b,~l,~p] | 3 7 5 4 6 | 1 2.3 1.6 1.3 2 |
| Residues in disallowed regions | 5 3 10 4 6 | 1.6 1 3.3 1.3 2 |
| Number of non-glycine and non-proline residues | 306 307 306 303 303 | 100% for all strains |
| Number of end-residues (excl. Gly and Pro) | 2 2 2 2 2 | |
| Number of glycine residues (shown as triangles) | 21 22 23 25 25 | |
| Number of proline residues | 12 10 10 11 11 | |
| Total number of residues | 341 341 341 341 341 |
[Abbreviations used in the Table:
Lbrzl -> Leishmania braziliensis; Lmjr -> Leishmania major;
Linf -> Leishmania infantum; Lmx -> Leishmania mexicana
Ldv -> Leishmania donovani]
Table 3: Referring to Ramachandran Plots of LPG2 protein of five different strains of Leishmania.
The models of different Leishmania strains were based on template PDB coordinates of 2i68, 1ee4, 1pw4, 1ej1, 3b5d and 1xm9. The PDB ‘2i68’ codes for transmembrane domain of the multi drug resistance antiporter from E. coli Emr E. This protein has antiporter activity and belongs to a family of plasma membrane proteins and proteins integral to membrane. The PDB ‘1pw4’ codes for protein with transporter activity and belongs to MFS general substrate transporter fold and is involved in glycerol metabolic process and glycerol 3-phosphate transport. It is a protein of plasma membrane and protein integral to membrane. The PDB‘1xm9’ codes for structure of armadillo repeat domain of plakophilin 1, belongs to all alpha proteins with alpha alpha superhelix fold which has ARM repeat forming plakophilin 1 domain . This protein is involved in molecular functions like signal transduction, cell adhesion and protein binding. The PDB ‘3b5d’ codes for x-ray crystallographic structure of Emr E multidrug transporter in complex with TPP, involved in antiporter like transporter function. It is a plasma membrane component. Few drugs have been targeted to this protein like dapsone and tamoxifen. The PDB coordinates of ‘1ee4’ code for crystal structure of yeast karyopherin (importin) in a complex with a c-myc NLS peptide. This karyophilin alpha protein have armadillo domain with ARM repeat. This protein is classified as transporter protein which is a transmembrane protein with transporter activity involved in import into nucleus, protein targeting to membrane and intracellular transport. This is a leucine rich alpha helical protein. The PDB coordinates of ‘1ej1’ code for crystal structure of mRNA 5’cap binding protein (eIF 4E) bound to 7-methyl-GDP. This protein is having RNA binding and translation factor activity and belongs to class of alpha and beta proteins. From all these template analyses, it is found that except ‘1ej1’, all other templates are transporter–like protein. LPG2 protein also belongs to this group of protein
It is found that the best model of LPG2 protein of all these strains consisted of only one chain (Table 2). In all the models 68-73 percent is helical. The best model of LPG2 protein of L. donovani is having highest number (24) of helices where minimum five and maximum sixteen residues take part in formation of a helix. In L. donovani three 310 helices are present. In L. mexicana 28 residues has been found to be forming a helix and helices in this strain accounts for 73% of total residues.
Functional Assignment of LPG2 Protein by SVM
From the comparative analysis of LPG2 protein functional assignment of five Leishmania strains shows that it belongs to transporter group of proteins (Table 4). LPG2 protein also belonged to trans-membrane region protein. LPG2 protein of L. mexicana strain M379 belonging to copper binding (58.6%), magnesium binding (58.6%), metal-binding, iron-binding (73.8%) and sodium-binding (78.4 %) protein function families. It also belongs to type II (general) secretory pathway (IISP) family and major facilitator family (MFS) (58.6%). LPG2 protein of L. braziliensis also belongs to incompletely characterized transport systems - putative uncharacterized transport proteins (73.8%), G protein coupled receptors (58.6%) and major facilitator family (MFS) (58.6%). From NCBI, it is also known that this protein is a nucleotide sugar transporter protein which is also likely to be post-translationally modified and belongs to chaperone or intracellular trafficking group of proteins.
| Functions from NCBI | Friedlin_ L. major _CAJ08033 |
L. donovani_ Sudanese1S_ AAC46914 |
MNYC/BZ/62/M379_ Belize country L. mexicana |
MHOM/BR/75/M2904_ L. braziliensis |
L. infantum |
|---|---|---|---|---|---|
| Nucleotide-sugar transporter | TC 2.A. Electrochemical Potential-driven transporters - Porters (uniporters, symporters, antiporters) | TC 2.A. Electrochemical Potential-driven transporters - Porters (uniporters, symporters, antiporters) | TC 2.A. Electrochemical Potential-driven transporters - Porters (uniporters, symporters, antiporters) (99.2%) | TC 2.A. Electrochemical Potential-driven transporters - Porters (uniporters, symporters, antiporters) | TC 2.A. Electrochemical Potential-driven transporters – Porters (uniporters, symporters, antiporters) |
| Transmembrane | Transmembrane | Transmembrane | Transmembrane | Transmembrane | |
| 1.Posttranslational modification 2.protein turnover |
Sodium-binding (78.4 %) | TC 9.B. Incompletely Characterized Transport Systems - Putative uncharacterized transport proteins (73.8%) | |||
| Iron-binding (58.6%) | Iron-binding (73.8%) | Iron-binding | |||
| Metal-binding | G Protein Coupled Receptors (58.6%) | ||||
| TC 2.A.1 Major facilitator family (MFS) (58.6%) | TC 2.A.1 Major facilitator family (MFS) (58.6) | ||||
| chaperones / Intracellular trafficking and secretion |
TC 3.A.5 Type II (general) secretory pathway (IISP) family | ||||
| Copper binding (58.6%) | |||||
| Magnesium binding (58.6%) |
Table 4: Comparative analysis of functional assignment of LPG2 protein in various Leishmania strains by SVMProt.
Various post-translational modification sites for LPG2 protein of various Leishmania strains were identified using protein predict program. Two aspargine glycosylation sites (at 2nd amino acid [NHTR] and at 335th amino acid [NDTS]) have been found in L. infantum and L. donovani whereas only one glycosylation site at 2nd amino acid [NHTR] was found in other three Leishmania strains (Table 5). Two protein kinase C activation sites were predicted in three Leishmania strains and one PKC site was predicted in other two strains. Four casein kinase II phosphorylation (CKP) sites are present in two Leishmania strains whereas in other strains one or two CKPs are present. One to five numbers of N-myristoylation sites are present in different Leishmania strains. Only one disulfide bond between 20th and 244th amino acid residues was detected in this protein of differentLeishmania strains (Table 5).
| Results | L. donovani | L. mexicana | L. major | L. infantum | L. braziliensis |
| ASN_GLYCOSYLATION (N-glycosylation site) N[^P][ST][^P] | Pattern: 2 à NHTR 335à NDTS | Pattern: 2 à NHTR | Pattern: 2 à NHTR | Pattern: 2 à NHTR 335à NDTS | Pattern: 2 à NHTR |
| PKC_PHOSPHO_SITE ( Proteinkinase C) [ST].[RK] | Pattern: 91 à SMK 337 à TSK | Pattern: 91 à SMK 337 à TSK | Pattern: 91àSMK | Pattern: 91àSMK 337àTSK | Pattern: 91àSMK |
| CK2_PHOSPHO_SITE ( Casein kinase II phosphorylation site) [ST].{2}[DE] | Pattern: 6 à SVME 308 à SDTE 320 à TTAE 338 à SKSE | Pattern: 6 à SVME 338 à SKSE | Pattern: SDLEà 308 | Pattern: 6à SVME 308àSDTE 320àTTAE 328àSKSE | Pattern: TDAEà 308 |
| MYRISTYL (N-myristoylation site) G[^EDRKHPFYW].{2}[STAGCN][^P] | Pattern: 140 àGSLLGA 155 àGLVWTF 178 àGSVSNS 283 àGIMIAL | Pattern: 140à GSLLGA 155à GLVWTF 178à GSVSNS 283à GILIAL 336à GTSKSE | Pattern: 178àGSVSNS | Pattern: 140àGSLLGA 155àGLVWTF 283àGIMIAL | Pattern: 178àGSVSNS |
| DISULFIND | 20-244 (length- 224aa) | 20-244 (length- 224aa) | 20-244 (length- 224aa) | 20-244 (length- 224aa) | 20-244 (length- 224aa) |
| Predicted secondary structure | Hà55.72% E à12.02% L à 32.26% |
Hà56.01% Eà10.85% Là33.14% |
|||
| Globularity | nexp = 91 (number of predicted exposed residues) nfit = 140 (number of expected exposed residues diff = -49.00 (difference nexp-nfit) So , protein not be globular | nexp = 87 (number of predicted exposed residues) nfit = 140 (number of expected exposed residues diff = -53.00 (difference nexp-nfit) So , protein not be globular |
Table 5: It shows comparative analysis of different motifs of LPG2 protein of five Leishmania strains. The motifs were predicted by Predict Protein server.
ELM server detected short functional sites in LPG2 protein. In L donovani, L. major and L. infantum, phosphothreonine motif binding a subset of FHA domains having a preference for an acidic amino acid at the pT+3 position (Nucleus, Replication fork) (LIG_FHA_2) has been predicted at 318-324[GKTTAES] position. In L. braziliensis the predicted site and amino acids involved were different (Site 306-312 and sequence is SATDAEN) (Table 6). Since phosphothreonine motif of LPG2 protein of L. braziliensis is different, according to this motif, LPG2 protein of all other four Leishmania strains are homologous to each other which is also confirmed by Clustal W and phylogenetic analysis. From analogy point of view, all the five strains are equal as the phosphothreonine motif is found in all five Leishmania strains.
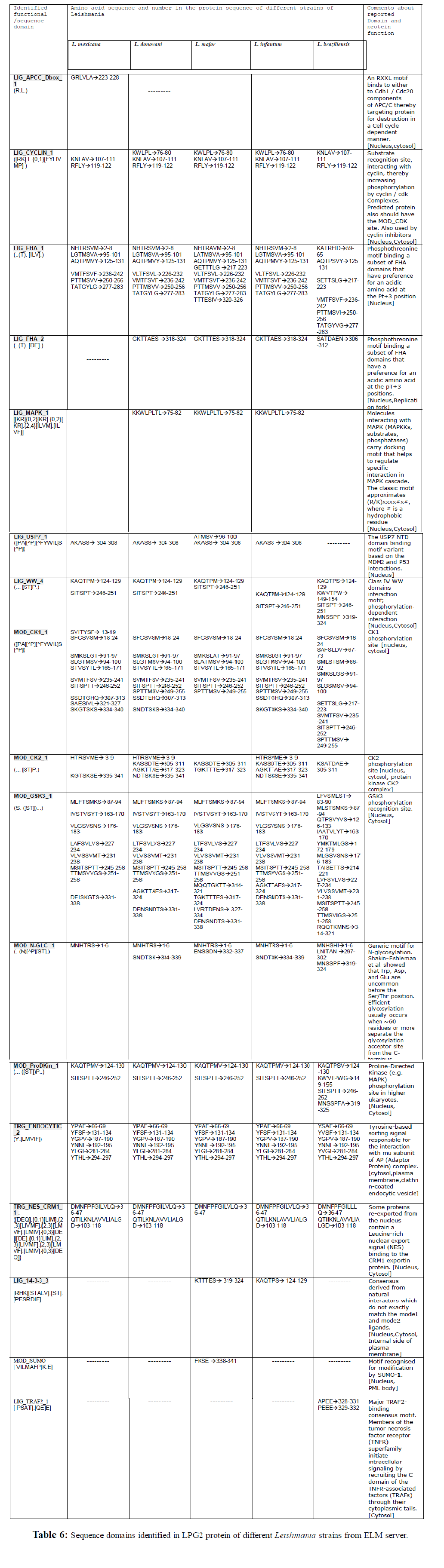
The MAP kinase (MAPK) cascades convey a signal in form of phosphorylation events. MAPKs are phosphorylated by MAP kinase kinases (MAPKKs), phosphorylate various targets, such as transcription factors and MAPKactivated protein kinases (MAPKAPKs), and are dephosphorylated and inactivated by several MAPK-phosphatases (MKPs) (Sturgill et al., 1991; Ahn et al., 1992; Nishida et al., 1993; Marshall et al., 1995). In LPG2 protein of threeLeishmania strains, MAPK interacting molecules (e.g. MAPKKs, substrates, phosphatases) carrying docking motif helping to regulate specific interaction in the MAPK cascade, have been detected in 75- 82 residues (Table 6).
Cell cycle depends upon the well orchestrated activation and deactivation of cyclin-dependent kinases which phosphorylate a number of substrates required for entry into the next phase of the cell cycle (Takeda et al., 2001). Three substrate recognition sites have been identified in LPG2 protein of Leishmania strains that interact with cyclin and thereby increase phosphorylation by cyclin / cdk complexes which are required for cell cycle events. Predicted proteins also have the MOD_CDK sites which are used by cyclin inhibitors (Table 6).
USP7 plays an important role in regulating cell proliferation and apoptosis through p53 and Mdm2 interactions (Saridakis et al., 2005). Motif containing amino acid sequence[AKASS-304th -308th] of LPG2 protein of fourLeishmania strains is the USP7 NTD domain binding motif variant based on the MDM2 and P53 interactions, this site is absent in Leishmania braziliensis (Table 6).
WW domain is one of the domains mediating cellular processes which require physical interactions between proteins (Bedford et al., 1998). Two to four numbers of class IV WW domains interaction motifs (phosphorylation-dependent interaction motifs found in both nuclear and cytosolic proteins) are present in LPG2 protein of Leishmania strains (Table 6).
CK1 is a ‘‘phosphate-directed’’ protein kinase which is able to phosphorylate with high efficiency Ser/Thr residues specified by a prephosphorylated side chain (either pS or pT) at position n–3 (or less effectively n–4) (Meggio et al., 1979; Donella-Deana et al.,1985; Flotow et al., 1990). This observation led to the concept of ‘‘hierarchical phosphorylation’’ (Roach et al., 1991) and the term ‘‘primed phosphorylation’’ to indicate the ability to phosphorylate residue(s) specified by another phosphorylated residue at a predetermined (critical) position. This feature is shared by a small number of acidophilic Ser/Thr kinases, notably CK1 and CK2, glycogen synthase kinase 3 (GSK3), and the Golgi apparatus casein kinase (G-CK) (Pinna et al., 1996). Casein kinase I have a wide variety of substrates. CK1 phosphorylation motifs (7-11 numbers) (throughout the LPG2 sequence) have been identified in LPG2 protein of Leishmania strains. Reports on various CK1 inhibitors are known which can be applied to LPG2 protein by docking to find novel drug candidates for leishmanisis treatment (Table 6) (Rena et al., 2004). Protein kinase CK2 is a pleiotropic and ubiquitous serine or threonine kinase, which is highly conserved during evolution (Faust et al., 2000). Protein kinase CK2 can phosphorylate many protein substrates in addition to casein. CK2 phosphorylation motifs have been identified in LPG2 protein of Leishmania strains, one at the beginning of amino terminus and others towards carboxy terminus.
Glycogen synthase kinase 3 (GSK3) is a well conserved serine/threonine kinase that is implicated in different cellular processes controlling cell proliferation and programmed cell death (Frame et al., 2001). In resting cells, GSK3 is a constitutively active kinase that phosphorylates a wide range of protein substrates to directly inhibit their biochemical activities, interfere with their sub-cellular localization, or promote their degradation (Ali et al., 2001). GSK3 phosphorylation recognition sites are found in LPG2 protein throughout the sequence of Leishmania. On comparison of GSK3 sites, amino acids involved in formation of this recognition site are same but in few cases it is different.
N-glycosylation motifs have been detected in different strains at the beginning of N-terminal and towards carboxy terminus. In LPG2 protein of L. braziliensis, three Nglycosylation motifs are present. Among the different types of glycosylation, the N-linked attachment of sugars to the polypeptide backbone is by far the most abundant modification (Vijay et al., 1998).
Two proline-directed kinase (e.g. MAPK) phosphorylation sites (124-130 and 246-252) are found in four Leishmania strains whereas in L. braziliensis, four PDK phosphorylation sites are present. One way that they achieve this is through direct interactions with substrate residues flanking the phosphorylation (P) site (Pinna et al., 1996; Lu et al., 2002; Songyang et al., 1996). The preference for proline at the P+1 position may be linked to downstream signaling mechanisms mediated by Pin1 proline isomerization (Zhou et al., 1999).
Tyrosine-based sorting signal sequences have been found throughout the LPG2 protein sequence of Leishmania which is responsible for the interaction with mu subunit of AP (adaptor protein) complex. Multiple sorting steps within eukaryotic cells are mediated by tyrosine-based sorting motifs. These motifs are recognized by the medium-chain subunits of heterotetrameric adaptor complexes (Stephens et al., 1998).
Some proteins re-exported from the nucleus contain a leucine- rich nuclear export signal (NES) binding to the CRM1 exportin protein. CRM1 mediates the nuclear export of proteins exposing leucine-rich nuclear-export signals (NESs) (Kutay et al., 2005). Two such motifs [36th - 47th and 103rd -118th amino acids] are detected in LPG2 protein of Leishmania strains except L. major where only one site [36th - 47th amino acids] is present. Hence the LPG2 protein manufactured in nucleus is transported to cytosol or Golgi due to presence of these motifs in LPG2 protein itself.
Interestingly, the sequences “KTTTES” and “KAQTPS” of LPG2 protein of L. major and L. infantum respectively matches a 14-3-3 interaction consensus site derived from natural interactors. Dimerization of 14-3-3 is essential for a good interaction (Cahill et al., 2001; Tzivion et al., 1998). P53 tumor suppressor protein, which plays a major role in maintaining genomic stability, has only one 14-3-3 binding site (Waterman et al., 1998). Hence LPG2 protein of Leishmania may have genomic stability role, which is required to be proved experimentally.
Only novel motif [FKSE] recognized for modification by SUMO-1 has been identified in LPG2 protein of L. major. The small, ubiquitin-related protein SUMO-1 is highly conserved from yeast to humans (Muller et al., 2001) and has been associated with subnuclear localization of many cellular proteins. Like ubiquitination, sumoylation leads to attachment of SUMO-1 to target proteins through the γ-NH2 group of lysine residues, using a cascade of E1, E2, and E3 enzymes. SUMO-1 is an important determinant of protein localization, required for the speckled nuclear distribution of the proteins PML, TEL, and HIPK2.
Likewise major TRAF2-binding consensus motif found in members of the tumor necrosis factor receptor (TNFR) superfamily, which initiate intracellular signaling by recruiting the C-domain of the TNFR-associated factors (TRAFs) through their cytoplasmic tails, is also present in LPG2 protein of only one strain i.e. L. braziliensis.
In this analysis we have identified many motifs in LPG2 protein of Leishmania, which were previously reported in other proteins of different organisms, i.e. yeast to mammals. It is learned from this computational analysis that in LPG2 protein of L. braziliensis several distinct motifs are present which are not found in other Leishmania strains. The graphical view of all the motifs that present in LPG2 protein of all Leishmania Strains are shown in (Fig. 5).

Figure 5: This graph shows involvement of different conserved motifs in LPG2 protein of different Leishmania strains from ELM Server. Different colors have been used to distinguish the presence of various motifs in different Leishmania strains.
Protein Ligand Binding Site Analysis
Potential ligand binding sites in LPG2 protein of all Leishmania strains (L. donovvani, L. major, L. infantum, L. mexicana and L. braziliensis) have been found by using pocket finder program (Table 7) (Ruppert et al., 1997). Different LBSs of LPG2 protein of Leishmania are shown in (Fig. 6). First LBS of all the strains involve more than 35 amino acids. At most thirty eight amino acids (L. mexicana) involved in formation of binding sites have been found and in few sites a single amino acid is changed or lacking in formation of ligand binding sites, have been detected. Lowest number (four) of amino acids involved in formation of LBSs has been found in L. infantum (7th). It is known from this analysis that some LBSs are similar in all the strains but other LBSs are specific to each strain. All LBSs of L. donovani and L. mexicana are very similar, except at 1st and 6th where one amino acid is absent in L. donovani (1st site: G-188th and 6th site: V-328th).
| Ligand Binding Site (LBSs) | Total number of amino acids in L. mexicana | Total number of amino acids in L. donovani | Total number of amino acids in L. braziliensis | Total number of amino acids in L. major | Total number of amino acids in L. infantum |
| Site 1 | 38 | 37 | 30 | 35 | 37 |
| Site 2 | 36 | 36 | 17 | 24 | 9 |
| Site 3 | 17 | 17 | 18 | 21 | 16 |
| Site 4 | 12 | 12 | 16 | 13 | 11 |
| Site 5 | 10 | 10 | 14 | 10 | 9 |
| Site 6 | 12 | 11 | 14 | 10 | 7 |
| Site 7 | 10 | 10 | 13 | 10 | 4 |
| Site 8 | 9 | 9 | 10 | 14 | 7 |
| Site 9 | 9 | 9 | 14 | 8 | 8 |
| Site 10 | 10 | 10 | 13 | 7 | 7 |
Table 7: It shows various possible Ligand Binding Sites (LBSs) and total number of amino acids and their positions that involved in each binding site of L. mexicana, L. donovani, L. braziliensis, L. major and L. infantum.

Figure 6: Prediction of Ligand Binding Sites (LBSs) of LPG2 protein of five Leishmania strains by the pocket finder program (a) L. donovani, (b) L. major, (c) L. infantum (d) L. mexicana and (e) L. braziliensis
All these structural, functional and proteomic analysis about LPG2 protein of Leishmania species will lead to identification of novel lead compounds to eliminate Leishmania infection in Bihar state, India.
Future Perspectives
As the structure of LPG2 protein is known from this study, novel lead compounds can be designed on the basis of ligand protein interaction (docking) scores of available antileishmanial drugs with LPG2 protein, defining the highest dockable compound and designing various analogues of the presently available drugs or defining a novel molecule on the basis of different binding sites of LPG2 protein of different Leishmania strains.
This study was supported by Indian Council of Medical Research (ICMR), Govt. of India. We are thankful to Dr. Meera Singh of ICMR for helping us during establishment of our division. We acknowledge Dr. Sindhu Prava Rana for helping us in preparation of the manuscript.