Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2017) Volume 6, Issue 1
Objective: The nucleoside transporter family is an emerging target for cancer, viral and cardiovascular diseases. Due to the difficulty in the expression, isolation and crystallization of membrane proteins, there is a lack of structural information on any of the mammalian and for that matter the human proteins. Thus the objective of this study was to build homology models for the three cloned concentrative nucleoside transporters hCNT1, hCNT2 and hCNT3 and validate them for screening towards the discovery of much needed inhibitors and probes.
Methods: The recently reported crystal structure of the Vibrio cholerae concentrative nucleoside transporter (vcCNT), has satisfactory similarity to the human CNT autologs and was thus used as a template to build homology models of all three hCNTs. The Schrödinger modeling suite was used for the exercise. External validation of the homology models was carried out by docking a set of recently reported known hCNT1 nucleoside class inhibitors at the putative binding site using induced fit docking (IDF) methodology with the Glide docking program. Then hCNT1 homology model was subsequently used to conduct a virtual screening of a 360,000 compound library and 172 compounds were obtained and biologically evaluated for hCNT 1, 2 and 3 inhibitory potency and selectivity.
Results: Good quality homology models were obtained for all three hCNTs as indicated by interrogation for various structural parameters and also external validated by docking of known inhibitors. The IDF docking results showed good correlations between IDF scores and inhibitory activities; particularly for hCNT1. From the top 0.1% of compounds ranked by virtual screening with the hCNT1 homology model, the 172 compounds selected and tested for against hCNT1, hCNT2 and hCNT3, yielded 14 new inhibitors (hits) of (i.e., 8% success rate). The most active compound exhibited an IC50 of 9.05 μM, which shows a greater than 25-fold higher potency than phlorizin the standard CNT inhibitor (IC50 of 250 μM).
Conclusion: We successfully undertook a homology modeling and validation for all human concentrative nucleoside transporters (hCNT 1, 2 and 3). The proof-of-concept that these models are promising from virtual screening to identify potent and selective inhibitors was also obtained using the hCNT1 model. Thus we identified a novel potent hCNT1 inhibitor that is more potent and more selective than the standard inhibitor phlorizin. The other hCNT1 hits also mostly exhibited selectivity. These homology models should be useful for virtual screening to identify novel hCNT inhibitors, as well as for optimization of hCNT inhibitors.
<Keywords: Concentrative nucleoside transporter homology modeling; Docking; High throughput virtual screening; Anticancer cancer agents; Antiviral agents; Hit enrichment
Human nucleoside transporters (NTs) belong to two families namely the SLC28 family of cation-linked human concentrative nucleoside transporter (hCNT) proteins (hCNT1, hCNT2 and hCNT3) and SLC29 energy-independent human equilibrative nucleoside transporter (hENT) proteins (hENT1, hENT2, hENT3 and hENT4) [1,2]. CNTs mediate the unidirectional cellular flux of natural nucleosides as well as nucleobase- and nucleoside-derived drugs used in cancer and anti-viral chemotherapy [3,4]; most of which are hydrophilic in nature, and need assistance to cross cell membranes. Thus, these transporters are key determinants of tissue distribution and cellular response to the drugs. Unlike, ENTs, which transport a broad range of substrate, CNTs have much narrower and differing substrate specificities. hCNT1 generally transports pyrimidine nucleosides, but also transports adenosine, hCNT2 is generally selective for purine nucleosides but also transports uridine, whereas hCNT3 transports both purine and pyrimidine nucleosides [5]. NT inhibitors, by regulating nucleoside concentrations endogenously, such as the concentrations of adenosine, can modulate their physiological processes, which might lead to therapeutic benefits. Thus, the development of novel nucleoside transporter inhibitors may play a significant role in the development of therapies for several diseases and disorders [3].
Transporter proteins belong to the membrane protein family and due to the difficulties in expression and isolation, there is little x-ray crystallographic structural information on these proteins and thus only a relatively small number of their structures have been deposited in protein data bank (PDB), making it challenging to build homology models of these types of proteins due to a lack of suitable templates. Recently, the first crystal structure of a Vibrio cholerae concentrative nucleoside transporter vcCNT was published at 2.4 Å resolution [6]. We found that the vcCNT crystal structure has satisfactory structural relationships with the human autologs, hCNTs, to allow for the building of hCNT homology models. In the present work, we developed homology models for all three cloned hCNTs. These new homology models were refined and validated by flexible docking of known ligands [7] (Figure 1). In addition, we also used the hCNT1 homology model to identify novel non-nucleoside hCNT1 inhibitors that are more potent than the standard hCNT1 inhibitor phlorizin, which will serve as lead compounds for developing new inhibitors and probes for studying hCNT1 biology as well as its therapeutic target potential. The models should also have utility for structure-based optimization of selective hCNT inhibitors [8].
Homology modeling
Homology modeling was performed using the Prime 3.1 module implemented in the modeling Schrodinger suite 2012 [9,10].
Template identification: The input sequences of hCNTs were obtained from the Universal Protein Resource (http://www.uniprot. org). BLAST [11] was then used to find homologous protein structures in the PDB database, using BLOSUM62 for similarity analysis. The crystal structure of the Vibrio cholerae concentrative nucleoside transporter (PDBid: 3TIJ) [6] in the PBD was identified as a potential template. Other, vcCNT pdbs structures were published later by the same group [12]. All three hCNTs showed identities above 35% compared to the 3TIJ vcCNT structure, which was considered good for building homology models (the rule of thumb being that a sequence homology of 30% or above is sufficient). The detailed identity, pertinent residues and scores are given in Figure 2.
Target template protein alignments: Prior to sequence alignment, the protein preparation wizard was used to check for errors in template PDB structure (3TIJ). Next, the globally conserved residues were identified using the Prime STA program. Secondary structure predictions were subsequently performed and then alignment was carried out using the Prime module (Figure 2).
Model building: After alignment with the template, the test sequence was further subjected to model building using energy based methods. The crystal bound ligand uridine (URI) and bound sodium ion was also included in building the models.
Model refinement: In the final stage, the homology model was built using four steps (i) copying of backbone atom coordinates for aligned regions and side chains of conserved residues, (ii) optimization of side chains, (iii) minimization of non-template residues, and (iv) building of insertions and closing of deletions in the alignment. The models were then subjected to refinement starting with the non-template loops using the ultra-extended sampling method with all other default parameters intact. The model was then checked for any missing side chains; and finally minimized using an all-atom minimization technique. The overlay of the homology models of the three hCNT1 are shown in Figure 3.
Model validation: The refined models were validated for their structural quality by Ramachadran Plots using the Molprobity program [13] (Figure 4).
The usefulness of the homology models were assessed by docking a series of known active compounds into the uridine binding site.
Ligand preparation
All docking ligands were taken from the literature [7], drawn within Maestro 9.3 using Builder, and converted to 3D structures. The ligands were further subjected to refinement in the Ligprep2.5 module. Ligprep makes ligands ready for docking by checking for errors and generating low energy conformations.
Protein preparation
The refined protein model was prepared using the protein preparation wizard in the Schrodinger modeling suite. It checks and corrects the protein model for missing side chains and loops, as well as bond orders. It was also used to assign protonation states of amino acid residues and to optimize the placement of hydrogen atoms, and the restrained minimization of all atoms to ready the protein for docking studies.
Extra precision (XP) docking
The refined homology models were used to generate docking grid files. Amino acid residues within a box of 15Å around uridine binding site with uridine removed were kept in the grid file and used for docking. The number of initial ligand poses was set to 10,000 and the best 1,000 poses were energy minimized within the binding site.

Figure 1: Structures of known nucleoside analogue hCNT inhibitors used in further validation of the homology models.

Figure 2:The sequence alignments between hCNTs and vcCNT (sequence identities are: hCNT1 36%; hCNT2 37%; hCNT3 39%). Red color indicates identical residues, orange color indicates similar residues and black color indicates important amino acids at vcCNT binding site.
Induced fit docking
In recent usual docking methods, the receptor is held rigid and flexibility allowed for the ligand. This can lead to misleading results, as in real life situations the proteins are flexible and undergo side chains and back bone movements [9,10,14]. Thus, an induced-fit docking protocol was used to account for flexibility in the receptor site. Residues and side chains around 10Å were kept flexible during the docking. The induced fit docking score (IDF score) is a sum of the Glide docking score and 5% of the prime energy from the refinement step. The IDF scores obtained were plotted against hCNT inhibitory activity (Figure 5), whereby the IC50 values were converted to pIC50 values to derive dependent values for the plot. To validate the poses of the docked ligands, we compared the pose of compound MeThPmR from the validation compound set, with the recently published crystal complex poses of substrates in the PBDid: 4PD6 and PBDid: 4PD8 structures, which are uridine and pyrrolo-cytidine, respectively. The overlays are shown in Figure 6.
Virtual Screening
XP docking for virtual screening was undertaken of a 360,000 proprietary compound library from the University of Cincinnati Children’s hospital using the hCNT1 homology model. The virtual screening workflow used to filter the compounds is shown in Figure 7.

Figure 3:Overlay of the homology models of hCNT1 (green), hCNT2 (turquoise) and hCNT3 (red). The uridine pose in the nucleoside binding site of hCNT1 model is shown in green sticks, while the major binding site residues (amino acids within 3 Å radius) with which it interacted are shown in green wires and labelled.

Figure 4:Ramachandran plots of homology models. (A) hCNT1, (B) hCNT2 and (C) hCNT3. Favored (98%) regions are demarkated by blue lines, and allowed regions by purple lines. Good amino acids are denoted by black circles and outlier amino acids are denoted by magenta circles.
Cell lines expressing hCNT1 or hCNT2
We clone and stably expressed and functionally characterized individually hCNT1 and hCNT2 proteins [15] in the nucleoside transporter deficient (PK15NTD) cell line provided to us by Dr. Ming Tse of Johns Hopkins University [16]. We also obtained PK15NTD cells stably expressing recombinant hCNT3 from Dr. Ming Tse [16]. Thus we then had in hand cell lines expressing each individual hCNT protein, which we could then use to evaluate compounds inhibitory potencies and selectivity at the hCNT transporters.
Testing of compounds for hCNT1, hCNT2 and/or hCNT3 inhibition
Now that we had cell lines expressing individual hCNTs, i.e., hCNT1, hCNT2 or hCNT3 we use a uridine uptake assay for testing library compounds for hits. Since uridine is a universal permeant (substrate) for nucleoside transporters and is transported well by all three hCNTs [16], we tested the inhibition of [3H] uridine transport in the cells by the selected library compounds. According the Glide docking ranking and inspection for drug-likeness, we chose 172 compounds of the top docking compounds on hCNT1 model and tested them. The compounds were initially screened at 10 μM and compound HM50 (Figure 8), which showed the best inhibition and selectivity as an hCNT1 inhibitor was further tested in a dose-response assay.

Figure 5:Plots of pIC50 values against induced fit (IDF) docking scores of nucleoside analogue hCNT inhibitors docked in the homology models during the validation process.

Figure 6:Docking complex model hCNT1-MeThPmR (red) at the binding site (amino acids within 3 Å of bound uridine in hCNT1are displayed. Uridine is also overlayed on the pyrrolo-cytidine and adenosine of the PDBid 4PD8 (green) and PDBid 4PD9 (yellow) structures, respectively.
Nucleoside transporter inhibition assay
[3H]Uridine uptake assay for testing inhibitory activitiy: We used [3H]uridine because it is an excellent substratea for all three hCNTs. The method of Ward et al. [17] was used. Cells were maintained in Eagle’s minimal essential medium/Earle’s Balanced Salt Solution with 0.1 mM nonessential amino acids, 1 mM sodium pyruvate and 10% fetal bovine serum at 37°C in a humidified atmosphere of a mixture of 5% CO2 and 95% air. For uptake studies, cells were seeded at 5 ⊆ 104/well in 48-well plates two days before the experiments. To conduct the uptake assay, cells were washed with sodium-free buffer (choline chloride 120 mM, Tris-HCl 20 mM, K2HPO4 3 mM, Glucose 10 mM, MgCl2 1 mM, CaCl2 1 mM, pH7.4) and incubated with sodium-free buffer (choline chloride 120 mM, Tris-HCl 20 mM, K2HPO4 3 mM, Glucose 10 mM, MgCl2 1 mM, CaCl2 1 mM, pH7.4) or Na-containing buffer (NaCl 120 mM, Tris-HCl 20 mM, K2HPO4 3 mM, Glucose 10 mM, MgCl2 1 mM, CaCl2 1 mM, pH7.4) for 30 min. Then 1 μM of [3H]uridine (27.0 Ci/mmol) in sodium-free or sodium-containing buffer was added to each well and incubated for 2 min at room temperature. For inhibition assays, the cells were preincubated in sodium-buffer (NaCl 120 mM, Tris-HCl 20 mM, K2HPO4 3 mM, Glucose 10 mM, MgCl2 1 mM, CaCl2 1 mM, pH7.4) containing test compound for 15 min before addition of 1 μM [3H]uridine solution, and incubating for 2 min. Uptake was terminated by rapid aspiration of the incubation mixture and washing the cells three times with ice-cold PBS. The cells were then solubilized overnight in 300 μl of 5% Triton X-100 and 200 μl of cell lysate was counted for 3H content using a scintillation counter. Protein concentration of the cell lysates was determined using the BCA protein assay kit (Pierce, Rockford, IL).

Figure 7:The virtual screening work flow used in the study.

Figure 8:hCNT1, 2 and 3 inhibitory activities and selectivity of the 14 hits identified from the combined virtual screening and biological testing. Compounds HM1-HM152, were tested at a concentration of 10 µM, while phlorizin was tested at 250 µM its IC50 for hCNT1. The data are the mean ± sem for three experiments.
All uptake assays were carried out in triplicate, and experiments were repeated at least three times. The concentration of compounds that caused 50% inhibition of [3H]uridine uptake (IC50) was calculated using a nonlinear regression curve fitting method in the Prism program (Version 5, GraphPad, San Diego, CA).
Unlike, the ENT family of nucleoside transports for which several potent single to sub nanomolar level inhibitors and probes are available [16,17], CNTs have lagged woefully behind in terms of reported potent and transporter subtype selective inhibitors, let alone reporter probes; particularly hCNT1. This hampers their biological studies and potential targeting for therapeutic purposes; and that is the context for this study; to improve the selective inhibitor and probe landscape for hCNTs.
Homology models
The input hCNT sequences were searched to find homologous protein structures using BLAST; and this resulted in the protein databank (PDB) structure ID 3TIJ, a crystal structure of Vibrio cholerae CNT [6] in complex with uridine and sodium as a matching protein for all three hCNT sequences, with sequence identities of 36, 37 and 39% for hCNT1, hCNT2 and hCNT3, respectively; are considered satisfactory for building homology models. The quality of the homology models were evaluated from Ramachandran plots and analysis with the Molprobity software [13]. The models of all lack the first three TM domains (Figure 3). The Ramachandran plot data for all the three models showed more than 90% of backbone dihedral angles residing in the favored regions (Figure 4).
As shown in the Ramachandran plot Figure 4A, the hCNT1 model indicated that 93.9% (384/409) of were in the favored regions of the; 99.0% (405/409) of all residues in the allowed regions; and there were only four outliers Asp260, Ile349, Lys506, Gln509. For the hCNT2 model (Figure 4B), 93% (384/413) of all residues were in favored regions; 100.0% (413/413) of all residues were in allowed regions; and there were no outliers. For the hCNT3 model (Figure 4C), 94.7% (390/412) of all residues were in favored regions, 98.3% (405/412) of all residues in allowed regions; and there were 7 outlier residues, Ile371, Asn420, Ile530, His 531, Leu532, Ile548 and Ile550. All the three homology models were thus found to be satisfactory for further structure-based design studies. We further validated the models by docking recently published known hCNT inhibitors on them [7] (Figure 1).
Binding site
When we compared the binding sites of hCNT1 with that of vcCNT, the shape of the active site cavity looks similar overall but their physicochemical characteristics varied widely. The important amino acid residues (Glu156, Glu332) from vcCNT were shown to have H-bond interactions with the uridine substrate bound at the active site. Similar interactions were observed with Glu321, Glu497 from hCNT1. The binding sites of hCNT2 and hCNT3 were bigger in comparison to that of hCNT1. The hydrophobic characters of the hCNT2 and hCNT3 binding sites appeared higher than that of hCNT1. A detail of the binding site, in type of atoms is given in Table 1.
Docking studies
To study the binding mode of hCNT inhibitors and gain structural insights in the binding site of hCNTs the homology models were subjected for docking studies using six known active ligands [7]. Initial XP docking studies showed all ligands were able to dock correctly at the binding site, but when the XP when the XP docking score was plotted against the pIC50 values, we found no correlation between them. However, when we used induced fit (IDF) docking, the protein-ligand interaction energies in the form of showed IDF scores (Table 2) showed a correlation with the pIC50 values (Figure 5). It should be noted that the hCNT1 model afforded the best correlation between the biological activity and the IDF docking score. Interestingly, the nucleoside ligands that we used in the docking were also shown to be selective towards hCNT1 in particular by Damaraju et al. [7]. Thus, the fact that hCNT1 model performed the best, is a further strong support for the good quality of the homology models; giving confidence these models will be useful in structure based design of selective inhibitors.
| Binding site atoms | Hydrophobic atoms | Side chain atoms | |
|---|---|---|---|
| hCNT1 | 180 | 36 | 153 |
| hCNT2 | 343 | 56 | 270 |
| hCNT3 | 257 | 56 | 216 |
Table 1: Binding site nature of the hCNT’s.
In the hCNT1 induced fit docking, the most active compound MeThPmRex exhibited a similar docking pose as uridine, with an IDF score of -888.06; and H-bond interactions were observed with polar side chains of Gln319, Asn496, Asn543 and Ser546. The furan ring engaged in aromatic pi-pi stacking interactions with Phe541. However, it lost a H-bonding contact with Glu321. When these results are compared with those of the less active compound MeFuPmR for to binding hCNT1, some interesting insights were gained. MeFuPmR has a lower IDF score of 885.96 that could be due to the loss of a polar contact with Ser546, and the change to pyrimidine nitrogen instead of oxygen atom in its interaction with Gln319. It also gained H-bonding contacts with Tyr357 as seen with uridine (Table 2).
| Inhibitor | IDF Score for hCNT1 | IDF Score for hCNT2 | IDF Score for hCNT3 |
|---|---|---|---|
| MeThPmR | -888.06 | -898.03 | -868.05 |
| ImPmR | -887.13 | -898.09 | -868.80 |
| MePrPmR | -887.14 | -898.32 | -871.21 |
| dMeThPmR | -885.38 | -896.65 | -869.92 |
| MeFuPmR | -885.96 | -895.52 | -866.72 |
| PrPmR | -886.32 | -898.09 | -869.93 |
Table 2: IDF docking scores for hCNT inhibitors (the IDF docking score is a sum of the Glide docking score and 5% of the prime energy obtained from the refinement step).

Figure 9:Dose-response curve for the most potent hit identified, compound HM50.
Virtual screening
Virtual screening is an important undertaking at the front end of high throughput screening (HTS) by wet lab assays that can often enrich hit numbers and significantly improve success rates. Structurebased design methods for virtual ligand screening have been successful in lead discovery due to the detailed structural information on the drug target. They are useful for understand ligand-protein interaction mechanisms as well. In the present work, we used the hCNT1 homology model in conjunction with a proprietary chemical database of over 360,000 compounds (maintained at the Cincinnati Children’s Hospital) to carry out virtually screening for novel inhibitors of hCNT1. We developed a protocol for the docking-based screening using a modified virtual screening protocol with hierarchical docking filters as presented in Figure 5, using the Schrodinger modeling suite 2012, and employing the Glide program for docking. A total of 172 compounds were selected according to docking score and drug-likeness for biological testing, with phlorizin being used as the standard hCNT inhibitor control, which was tested at 250 μM, its IC50 against hCNT1 [18]. Compounds were tested at 10 μM concentration, and were screened against all individual three hCNTs stably expressed in PKNTD cell lines [12-14]. After biological screening, we obtained 14 novel selective (Figure 8) non-nucleoside hCNT1 inhibitors (Table 3), an 8% success rate. We determined the IC50 of the most potent hit, compound HM50 to be 9.05 μM, much lower than that of the control inhibitor phloridzin (IC50 of 250 μM) whereas IC50 for phlorizin (standard control) as already mentioned was 250 μM (Figures 9 and 10).

Figure 10:Docking complex model of hCNT1-HM50 (purple) and overlayed pose on adenosine of 4PD9 (yellow). Amino acids within 3Å of docked HM50 are displayed.
| Name | Structure | hCNT1 % Inhibition | hCNT2 % Inhibition | hCNT3 % Inhibition |
|---|---|---|---|---|
| HM7 | 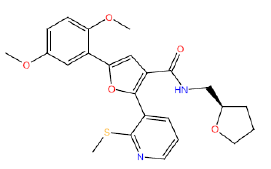 |
52 | 22 | -32 |
| HM11 | 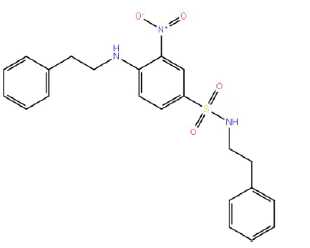 |
56 | 42.5 | -22 |
| HM21 | 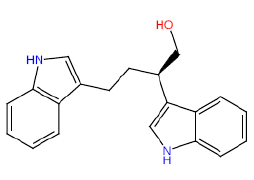 |
65 | 33 | 4 |
| HM25 | 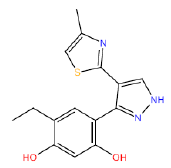 |
52 | 13 | -21 |
| HM50 | 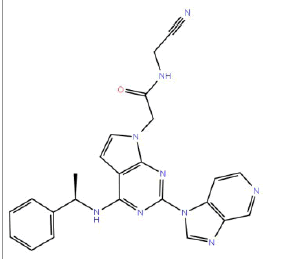 |
87 | 65 | 10 |
| HM58 | 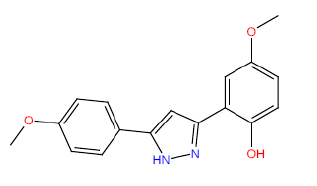 |
60 | 52.5 | -4 |
| HM59 | 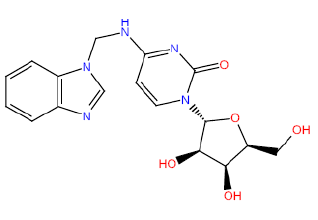 |
50 | 35 | 52.5 |
| HM64 HM64 | 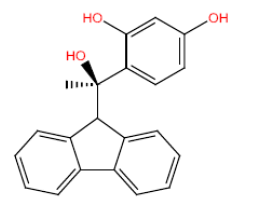 |
60 | 47 | -45.5 |
| HM66 | 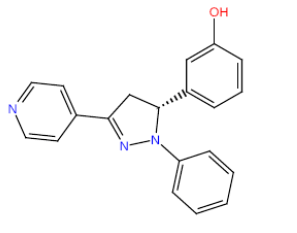 |
63 | 25 | 15.5 |
| HM68 | 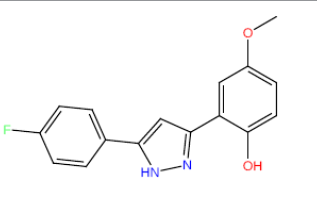 |
66.5 | 46 | 13.5 |
| HM70 HM70 | 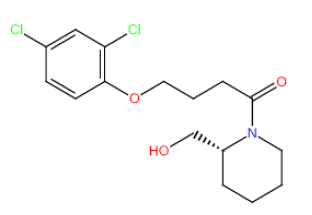 |
60 | 26 | -5.5 |
| HM72 | 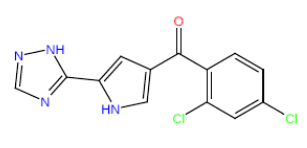 |
55.5 | 33 | 16 |
| HM85 | 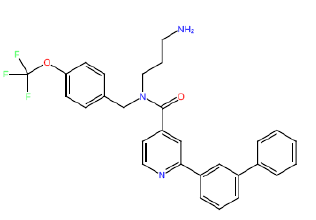 |
55 | 20 | -25.5 |
| HM152 | 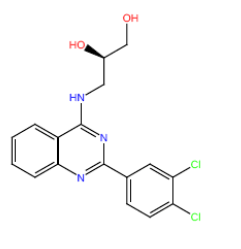 |
76.5 | 22.5 | 28 |
Table 3: Structures and hCNTs inhibitory activities of Hits obtained after biological screening at 10 µM concentration.
In conclusion, we have constructed homology models of all three human concentrative nucleoside transporters, hCNT1, hCNT2 and hCNT3. The homology models were refined and validated by standard protein validation tools as well as with induced fit docking of known ligands which were rank-ordered accordingly. The flexibility introduced by the induced fit docking protocol allowed us to obtain reasonable correlations between the binding affinity expressed as IC50 values and the docking energies. One of the most challenging problems in most current docking methods is the development of protocols that can rank compounds in order of biological activity based on scoring function predictions of binding affinity to proteins. Thus, for us to construct and validate homology models and use them to successfully dock and rank ligands to obtain reasonable correlations between IC50 values and docking scores, is a rare feat that serves not only as an example of how to address this challenging problem, but also provide useful models for the design of new hCNT inhibitors and optimization of novel lead molecules. Further we carried out virtual screening on the hCNT1 homology model to find novel inhibitors at a better success rate (8%) than usually obtained in HTS campaigns (~1%). Importantly, a novel hCNT1 inhibitor (HM50) was identified that is 25 times more potent than the standard compound phlorizin. The potent hCNT inhibitors have potential as probe tools for hCNT biology, and as potential lead compounds for therapeutic agents in various disease states [19-22].
Nucleoside transporter studies in our laboratory were supported in part by funds from NIH grant no. R01GM104503 and startup funds from Rosalind Franklin University of Medicine and Science awarded to JKB. The content is solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.