Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Review - (2024)Volume 17, Issue 2
Here we discuss the development of protein scaling theory, starting from backgrounds in mathematics and statistical mechanics and leading to biomedical applications. Evolution has organized each protein family in different ways, but scaling theory is both simple and effective in providing readily transferable dynamical insights, which are complementary for many proteins represented in the 60 thousand static structures contained in the online Protein Data Base (PDB). Scaling theory is a simplifying magic wand that enables one to search the hundreds of millions of protein articles in the Web of Science and identify those proteins that present new cost-effective methods for early detection and/or treatment of disease through individual protein sequences (personalized medicine). It complements and extends the most popular methods for studying protein evolution, based on amino acid sequence alignments. Applications include evolution of Covid and the abrupt end of the pandemic.
Critical point theory is general and recently it has proved to be the most effective way of describing protein networks that have evolved towards nearly perfect functionality in given environments (self-organized criticality). Evolutionary patterns are governed by common scaling principles, which can be quantified using scales that have been developed bioinformatically by studying thousands of PDB structures. The most effective scale involves hydropathic globular sculpting interactions averaged over length scales centered on membrane domain dimensions.
A central feature of scaling theory is the characteristic length scale associated with a given protein’s functionality. To gain experience with such length scales one should analyze a variety of protein families, as each may have several different critical length scales. Evolution has functioned in such a way that the minimal critical length scale established so far is about nine amino acids, but in some cases it is much larger. Some ingenuity is needed to find this primary length scale, as shown by the examples discussed here. Often a survey of the evolution of the protein suggests a means of determining the critical length scale.
Protein; Evolution; Protein data base; Scaling theory; Amino acid sequence
Before we discuss the mathematical and statistical basis for protein scaling, we should first consider the complexity of the problem; protein molecular complexity. A typical protein chain may contain 300 sites, each potentially occupied by one of 20 amino acids. The resulting number of possible amino acid sequences is 30020~1046, a number far larger than is normally encountered in physics, even in astrophysics. We shall later see bio-medically important examples where a single site amino acid mutation is enough to change success into failure. Is it possible to explain such sensitivity using fundamental principles? Perhaps it is, although every protein family is different.
Several ambitious statistical studies of protein sequences are available. A broad evolutionary (2001) study of 29 proteomes for representatives from all three kingdoms; eukaryotes, prokaryotes and archaebacterial, showed that proteins have evolved to be longer in eukaryotes, with more signaling heptad transmembrane helices in eukaryotes [1]. These are the signaling proteins which are the basis for about half of modern drugs. This very well-studied field is now classical (the Merck 106 sq. ft. Whitehouse Statn Research Lab specialized in these drugs and closed in 2014), while research on most other proteins has just begun. A sophisticated group wish list (>30 authors) of evolutionary problems associated with sequence evolution reported in (2012) that “current efforts in interdisciplinary protein modeling are in their infancy” [2].
Newtonian all-atom models are often unable to connect structural dynamics to function [3,4]. Evolutionary improvements are not easily recognized, as structures are seldom available for most species and backbone structural differences are often too small to explain evolutionary progression. For example, the chicken and human peptide backbone coordinates of lysozyme c, Hen Egg White (HEW) are indistinguishable, even at highest attainable X-radiation (X-ray) resolution in hundreds of PDB structures [5].
Science has grown rapidly in the last 60 years and the science curriculum has struggled to keep pace. Here we often use the word “classical” to indicate topics that are old enough to have gained wide acceptance and “modern” to describe the revolutionary changes that have taken place, often since ~2000. This review includes many modern ideas, which have enabled simple, powerful tools to organize molecular biology in new directions. The power of these tools will be illustrated by a few examples, especially involving evolution of protein functions.
Mathematical tools
Mathematical and scientific literature, as measured by numbers of papers published each year, is growing super-linearly, with the number of specialized journals proliferating in the 21st century. Citations are widely used as a measure of the impact of research efforts, if not the absolute importance of their content. This growth has stimulated more than 700 papers analyzing the literature scientometrically. The most spectacular scientometric study so far surveyed nearly all the 20th century literature, consisting of 25 million papers and 600 million citations, with 106-107 more entries than typical data bases and unique in the history of epistemology [6]. This study identified a citation transition, which occurred around (1960) and which is the earliest example of the cultural effects of globalization [7].
We can compare the development of different mathematical tools by using carefully chosen key words to search the Web of Science [8]. For example, although analysis is also a branch of mathematics, “analysis” is used in far too many non-mathematical contexts to be useful in a key word search. Useful mathematical words are algebra, geometry and topology; these yield surprising phase transitions over the last 30 years (Figure 1). It is plausible that the glasnost emigration of Russian computer scientists around (1990) facilitated development of many biological data bases and contributed to the success of the human genome project. Note not only the abrupt increase around 1990 associated with glasnost, but also the rapid increase in topology, which crosses over algebra with the advent of the internet in 2000. The fastest growing field, topology, is well suited to discussing protein network structure and evolution. One might have supposed that geometry, the oldest of the mathematical disciplines, would have stagnated by now, but modern geometry has evolved into differential geometry, a tool whose concepts again are well suited to discussing the functions of sculpted globular proteins. The two-year (1989-1991) jump of special functions, a subclass of analysis, by a factor of >20, is far larger than any of the general jumps in Figure 1. Other subclasses also show large 1989- 1991 jumps, for instance, “differential geometry”, a factor of 9 and “network topology”, a factor >20.

Figure 1: The annual numbers of papers on each of three mathematical disciplines, with the geometry number divided by 2. The entire geometry profile covers 330,000 papers. 
Statistical mechanics
In his enormously popular Dublin (1943) lectures and book, “What Is Life?”, Erwin Schrodinger proposed that we could progress in answering this question by using statistical mechanics and partition functions, but not quantum mechanics and his wave equation. He described an “aperiodic crystal” (today we would call it a glass) which could carry genetic information, a description credited by Francis Crick and James D. Watson with having inspired their discovery of the double helical structure of Deoxyribonucleic Acid (DNA) [9,10]. Schrodinger arrived at this picture from thermodynamic theories concerning protein stability and information content. He observed that proteins are not only exponentially complex, but also must be near thermodynamic equilibrium, as they function nearly reversibly. Thus one can say that Schrodinger may have been the first theorist to conjecture that protein functionality could be usefully described thermodynamically.
Given what we know today, Schrodinger might have started more simply with the isothermal curves of the van der Waals equation of state, from R L Rowley, Web Module; Van der Waals Equation of State, with author's permission. The equation was invented by van der Waals in 1881 (Figure 2). Apart from providing a quantitative description of the liquid-gas transition of many molecular liquids, this equation exhibits spinodal phase separation topped by a critical point on the critical temperature isothermal. The critical features of this model are common to many systems at or very near thermodynamic equilibrium. Specifically, one can imagine that protein functionality can involve transitions between two end states, called “open” and “closed” by Karplus in his protein example [3]. Also the two phases are either more stable or have faster kinetics. Optimization of both properties defines a functional critical point. Then if one knows from general considerations some quantitative properties of any protein sequence, one may be able to recognize extrema and critical points and quantify behavior in their neighborhood, without using elaborate Newtonian simulations. The comparative advantages of this more abstract method are its simplicity and universality, which makes it transferable. Such a method can easily treat evolutionary protein sequences by following their nearly critical properties and may also facilitate identifying essential features in any protein and relating them to its functionality.

Figure 2: Cubic isothermal curves for the van der Waals equation of state. For T>Tc, the curves are monotonic and contain no extrema. For T
The importance of critical points in statistical mechanics has led to many studies of their properties in toy models, especially Ising models, which are lattice models with only nearest neighbor interactions. Ising himself showed (1924) that his models for dimensionality d=1 do not contain a phase transition (a general property of all d=1 models) and was unable to persuade himself that a transition existed for d=2. L. Onsager studied critical point neighborhoods for a two-dimensional Ising model (1944), which he had solved exactly [11]. For d>4 the critical exponents of correlation properties are integers or half-integers (mean-field theory, used by L. Landau in his book on statistical mechanics). K Wilson solved the most difficult case, d=3, using the renormalization group methods of particle theory. The critical exponents of the d=2 and 3, Ising models are irrational sums of power series [12]. The Wiki discussion of Ising models emphasizes random walks, but evolution has converged to make proteins far from random, as we shall see quantitatively.
The existence of irrational exponents for d=2 and 3 toy model phase transitions is suggestive, because empirical power law fits (which appear to be linear on log-log plots) are common in engineering discussions of nearly optimized systems. Power laws describe self-similarity (a power of a power is a power) and self-similarity is an appealing way of fitting together proteins over a wide range of length scales. Mandelbrot discussed geometrical self-similarity in terms of fractal exponents and power-law iterations (Mandelbrot sets) [13]. Per Bak connected self-organized criticality to science and to evolution [14,15].
Scaling
The general ideas discussed in 2012 and similar subsequent discussions of evolutionary dynamics and function were unable to make contact with statistical mechanics and critical points [2,16,17]. General ideas are suggestive, but can they produce tangible biophysical and bio medically relevant results [18]? The positive answer to this challenge came from an unexpected direction, bioinformatic thermodynamic scales. The Kelvin temperature scale is central to entropy and general statistical mechanics and T=0 on the Kelvin scale corresponds to -273.16 °C. Protein globular shapes are determined by competing hydrophobic forces (pushing segments inwards towards the globular cores) and hydrophilic forces (pushing segments outwards towards the globular water interface). Moreover, the leading physicochemical properties determining protein mutations are hydrophobicity, secondary structure propensity and charge [19].
To quantify these effects in the classic period of biophysics (before 2000), no less than 127 hydropathicity scales were proposed. Each scale had its merits and was based on at most only a few dozen measurements. Few attempts were made to compare their accuracies or applicability to properties other than those used in their definitions [20]. Interscale correlations were typically only ~70%. Meanwhile, the number and accuracy of PDB structures had grown enormously, creating the opportunity to re-examine the early geometrical definitions based on average neighboring hydropathicity volumes or average surface areas in Voronoi partitions of proteins into amino-acid centered units with van der Waals radii [21-23]. Note that the combination of Stokes’ theorem and modern differential geometry suggests that there should be a close connection between the volume and surface definitions. The surface one in particular emphasizes hydrogen bonding to the weaker water films which have shaped globular proteins in evolution, much as rocky coastlines are shaped by the pressures of tidal water waves [13,24].
With the stage set, in (2007) Brazilian bioinformaticists Moret and Zebende (MZ) built the interdisciplinary bridge connecting proteins to statistical mechanics and critical points [25]. They evaluated Solvent-exposed Surface Areas (SASA) of amino acids in >5000 high-resolution (<2A) protein segments, a data base about 100 times larger than the data bases used for the classical scales. MZ fixed their attention on the central amino acid in each segment. The Length (L) of their small segments L=2N+1, variable (N), varied from 3 to 45, but the interesting range turned out to be 9 ≤ L ≤ 35. Across this range they found linear behavior on a log-log plot for each of the 20 Amino Acids (AA),
log SASA(L) ~ const - Ψ (aa) logL (9 ≤ L ≤ 35)
Here Ψ (aa) is recognizable as a Mandelbrot fractal. It arises because the longer segments fold back on themselves, occluding the SASA of the central aa. The most surprising aspect of this folded occlusion is that it is nearly universal on average and almost independent of the individual protein fold. Thus this striking universal result transcends and compresses thousands of individual protein folding simulations.
It is plausible that the MZ fractals exist because protein evolution has brought average protein SASA near critical points in each of the 20 AA subspaces. Remarkably, these subspaces span the same length frame 9 ≤ L ≤ 35, independently of whether the aa is hydrophilic (near the globular surface) or hydrophobic (buried in the globular interior). One cannot “prove” with mathematical rigor these connections, but one can test (prove) them in the context of the evolution and functionality of many protein families. Comparing the results with those obtained with the classical scales enables us to estimate the relative merits of various scales.
When an {X (aa)} scale is available, it can be shifted linearly to a new {Xʹ (aa)} scale
{Xʹ (aa)} = a {X (aa)} + b
Given the two constants a and b, one can arrange all scales to have the same average value and difference of largest and smallest values.
< {Xʹ (aa)} >=< {X (aa)} >, Xʹ (aal)}-{Xʹ (aas)} = X (aal)}-{X (aas)}
Moret and Zebende compared their fractal scale hierarchically to seven classical scales, the closest of course being, which utilized only the SASA of a single aa, averaged over a few entire protein structures available in (1985) [22]. Paradoxically, the MZ Mandelbrot fractals based on short segment log-log plots may be more significant and not just because of critical points in configuration space. Both protein dynamics (on a scale of ms) and protein evolution (on scales as short as thousands of years) are difficult to understand in the context of their ~1046 configurational complexity (Levinthal’s paradox) [26].
The MZ bioinformatics success could not have occurred in the classical period before 2000, because the classical structural data base was too small. The significance of protein sequence comparisons (alignments) was summarized at the end of this period by Rost, where he saw hints of what was to come [27]. At first, it was not obvious why the MZ success occurred, but retrospectively we can see how fractal segmental character implicitly includes evolutionary optimization through exchange of modular building blocks. Amino acid hydromodularity is apparently the best-documented example of effective parameter space compression [28].
Although is closest to MZ, it is convenient to compare results obtained from the MZ scale (which implicitly describes second-order phase transitions), with those obtained from first-order protein unfolding measured by enthalpy changes from water to air (1982 Kyte-Doolittle (KD) Ψ scale; this is also the most popular Ψ scale, which we call the standard scale) [22,29]. The differences between results obtained by the two hydropathicity scales should reflect improvements in accuracy, which could be bio-medically important. Complex protein interactions can even be dominated by first-order interactions for smaller sliding window length W and by second-order interactions at longer lengths (following section).
In a few cases hydropathic (inside/outside) Ψ shaping may not be the most important factor, in which case we can turn to secondary structure (inside/outside) propensities of α helices and β strands [30]. Hydrogen bonding is longitudinal for α helices and transverse for β strands, so we are not surprised to find that the (inside/ outside) differences are small for α helices and larger for β strands which can bind outside. Protein binding can involve the β strand exposed propensities for some cases. Another β strand scale was constructed from the core sequence of amyloid β and we will return to this later [31]. It is designed to emphasize aggregative “Hot Spot” propensities for binding on a length scale of 7 aa and it will be referred to as β Hot Spot (βHS).
At first it may seem complex to have two hydropathicity scales Ψ and two β strand scales. Still, by comparing not only the scales with each other (through their Pearson correlation functions r), but also their performances on protein regions known to be structurally important, we will gain thermodynamic insights into molecular binding otherwise unobtainable. The best part is that comparative calculations with these scales are transparent and extremely easy and can be implemented using only Excel spreadsheets and their included software subprograms.
Critical length scales
In normal liquids critical opalescence couples long wave length light waves to long wave length density fluctuations. Near a critical point could long wave length ice-like film waves (sometimes called “soft modes”) couple to long wave length solvent-exposed protein area fluctuations, thus explaining the origin of the MZ fractals? This is a difficult question. In the apparently simpler system of quenched (non-equilibrium) glasses there has been much theoretical discussion of the possibility of a diverging length scale at the glass transition, especially connected with long-range stress fields [32,33]. To discuss self-organization, phase diagrams and physical properties of network glasses, one must start from specifics of their chemical bonds, as well as both the local and extensive topological properties, which include stress percolation [34-36].
Here we argue that biomedically important results are obtainable by judiciously combining specific length scales W=2M+1 with one or more of the Φ={hydropathic Ψ/β strand scales}, denoted generically by Φ. Given Φ (aa), we calculate the modular average;
Φ (aa,W) = average(Φ (aa-M), Φ (aa+M))
which is a rectangular window from aa -M to aa +M. It is possible to iterate this process, for instance one can easily see that gives a triangular window extending from aa -2M to aa +2M. In practice for most cases rectangularly smoothed Φ (aa, W) appear to give best results. By using Φ scales we “dress” modular building blocks and enable protein interactions to appear to be short range with cutoffs.
Φ (aa,W,W) = average(Φ (aa-M,W), Φ (aa+M,W))
Given that Φ (aa, W) is a good variable, how do we determine W? Experience with many examples suggests that all protein families are different, because their functions differ. We reflect some of these differences by choosing an optimized W and others appear through comparisons between different components Φ. Because all four Φ scales have general meanings, comparison of Φ (aa, W) profiles often produces easily interpreted results. Far from being complex, the multiple tools associated with Φ (aa, W) are powerful aides for exploring protein complexity. They automatically incorporate universal aspects of globular evolution.
Hinges and pivots
Given a protein profile Φ (aa, W), one notices immediately that it has two kinds of extrema, hydrophobic maxima and hydrophilic minima. It is natural to suppose that the maxima act as pivots or pinning points for the conformational motion that is functionally significant, while the minima act as hinges. This language does not specify the conformational motion in Euclidean space that is functionally significant, as it jumps directly from the universal sequence geometries of Φ to function. What happens if we attempt to go elastometrically only between sequence and structure, by using the isotropic vibrational amplitudes of individual amino acids measured in structural studies?
The picture of differential aqueous sculpting of globular protein surfaces near a critical point can be compared to elastometric treatments of hinge-bending conformational transition pathways [15,37-39]. For some enzymes there are similarities in shape that correlate with functionality through correlations of flexible α helices [40]. This example is complementary to hydropathic shaping supplemented by exposed/buried β strand interactions. Although muscle contraction is mediated by a myosin cross-bridge which exists in two (open/closed) conformations, these were not explained by known conformers of myosin [41]. Instead there is an iterated hand-over hand motion of myosin along actin filaments [42].
Variance, correlations and level sets
One of Excel’s convenient software tools is variance, which can be used to quantify trends in extremal (phobic-philic) widths as functions of both the choice of Φ (for instance, Ψ KD verses Ψ MZ) and W. Informally variance is known as "mean of square minus square of mean" or,
Var(Φ (aa,W )) = Σ (Φ (aa,W)- < Φ (aa,W) > )² = Σ Φ (aa,W )² – n ( < Φ (aa,W ) > )²
where the sum is taken over n consecutive amino acid sites. In the context of the MZ Ψ scale, the variance measures the hydropathic roughness of the globular surface of the n sites of the protein chain segment. The local or global roughness can affect dynamical functions, which should occur neither too fast nor too slowly, in order to synchronize with other protein motions [43]. Mixtures of rougher granules have lower packing densities and the granular knobs can jam kinetics [44].
Variance is a useful quantity in studying protein evolution and dynamics because it combines extremal hydropathic pivots and hydrophilic hinges on an equal footing. One might suppose that such a simple function would have been used for long times in biology. In fact, its bioinformatic importance was first realized only in (1911) by R. Fisher (then a student), while its publication centenary was 2018. Fisher used it to describe “the correlation between relatives on the supposition of mendelian inheritance”. It conveniently represents the random combinations of parent genes.
Pearson correlations themselves are normalized cross variances between two functions, for instance two sequences of the same protein from different species or strains X and Y (-1 ≤ r ≤ 1).
r =< (X (aa)) − < X (aa>)Y (aa) − < Y(aa >>)/(<(X (aa))² > − < X (aa>²)1/2>) < (Y (aa))² > − < Y(aa >²)1/2
Level sets were developed to track the motions of continuum interfaces-applied here to protein globular surfaces [45]. Mathematically oriented readers will find “simple” explanations of their background and comparative computer science advantages online, for instance, under “Level set methods: An initial value formulation”. Practical applications of level sets have emphasized image analysis and have gradually evolved to include Voronoi partitioning, just as has been used for deriving protein hydropathicity scales since 1978 [46-48]. We expect, of course, that hydrophobic pivots move most slowly, while hydrophilic hinges move fastest. When there are two or more level pivots or hinges, it is likely that this is not accidental (nothing in proteins is) and we can test this assumption by comparing profiles with different scales, the KD Ψ and MZ Ψ scales, for example.
Evolution and mutated aggregation, HEW
Lysozyme c, aka HEW was for some time the most studied protein; the PDB contains more than 200 humans and 400 chicken HEW structures. HEW is also present in many other species, not only in the chicken sequence (domesticated from jungle fowl dating back 400 million years), but in most other vertebrates, almost unchanged in its peptide backbone structure. The backbone structure is exceptionally stable, with human and chicken C α positions superposable to 1.5A°, while the aa sequence mutates from chicken to human with 60% aa conservation, well above the 40% minimum usually necessary for fold conservation [10,49]. Comparison of Kappa caseins from mountain and domesticated goats suggests that chicken domestication of lysozyme c may have changed its aa sequence by ~2%.
HEW is a comparatively small 148 aa proteins, which has a nearly centrosymmetric tripartite α helices (1-56 and 104-148) and β strands (57-103) secondary structure. During its long career, HEW has performed at least three functions, as an enzyme, an antibiotic and an amyloidosis suppressor. The relative importance of these functions has changed from species to species and it seems likely that these changes are reflected in the amino acid mutations that have maintained the centrosymmetric structure.
Because we are most interested in the evolution of the properties of HEW from chickens to humans, we plot the Roughness (R) or variance (W) as RS (W)/RHuman (W) in Figure 3, using the fractal MZ Ψ scale, for a range of Species (S). There is a broad peak, together with a narrow peak, both centered on W=69, roughly half the protein length. The peak also occurs at W=69 with the KD Ψ scale, but its amplitude is only ~60% as large, so the roughness evolution is better described as thermodynamically second order. Similar narrow-broad peak structures have been observed in critical opalescence spectra, where the broad peak, associated with phonons, is called the Mountain peak [50]. Note that this “universal” peak applies to the terrestrial vertebrates, but not to zebrafish (Figure 3).

Figure 3: The roughness RS(W)/RHuman(W) ratios for hen egg white are shown for six terrestrial species. These ratios exhibit both a broad and narrow peak, both centered on W=69, suggesting that the evolution of Hen Egg White (HEW) from chicken to human has been directed towards improving a specific function, avoiding aggregation (amyloidosis). Note: R: Roughness factor; W: Variance; MZ: Moret and ![]()
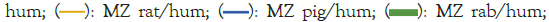
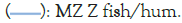
The structure-function relations giving rise to these peaks can now be profiled with W=69, as shown in Figure 4 for the extreme terrestrial cases of chicken and human. Note that normalizing RS (W) by RHuman (W) in Figure 3 is natural, because human structures have evolved to be closest to critical and smoother. This means that in most cases the critical limiting behavior of ideal functionality is nearly reached with the human sequence, described partly by RHuman (W). It is obvious from Figure 4 that on the W=69 length scale, which is approximately half the HEW length, the largest effect of evolution has been to stiffen the flexible central β strands by making them less hydrophilic.

Figure 4: Hydropathic profiles of human and chicken HEW, using the fractal Moret and Zebende (MZ) scale, as suggested earlier. Note the large stabilization by the human strain in the β strand region, compared to the small differences in the α helical regions. Here increasing ordinate corresponds to increasing hydrophobicity and increasing rigidity. The four numbered sites near the centre show large mutated aggregation rate changes. 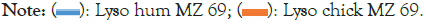
While the results shown in Figure 4 dramatically confirm the previously hidden content of HEW amino acid sequences, what is it? The characteristic length scale for amyloidosis is W=40, because this is the length of the Aβ fragment responsible for forming amyloid fibrils. On this length scale one can examine the effects of mutations on HEW aggregation rates and a detailed discussion shows consistent shifts [49]. Presumably amyloid suppression is a key function for advanced species with larger neuronal networks which must be stable for longer lifetimes.
One can also discuss mutated aggregation rates in a similarly centrosymmetric protein, measured and analyzed there using W=5 only (no other values of W were considered). The 98 aa α/β protein Acylphosphatase (AcP) resembles HEW (αβα) in that its sequence is still nearly Ψ centrosymmetric, but it is more complex with five regions instead of three (β<1-19>, α<20-32>, β<33-54>, α<55-70>, β<71-98>). A parameterized W=5 method for studying HEW mutations was applied to AcP, with disappointing results: increases in mutated aggregation rates were expected only in the central region and found only in the N- and C-terminal wings [19].
When the S/human roughness ratios are plotted for Acyl-1, the results are much more complex than in Figure 3 for HEW. The difference is attributed to its richer α/β structure [49]. The overall scale, as measured by chick/human, is about 30% enhancement, which is about 10 times smaller than that shown for HEW in Figure 3. Nevertheless, three features were easily identified. Chick/ human gave peaks at W=43 and W=25, as well as a human peak at W=13. These three values of W probably reflect interactions with three other (unknown) proteins. One can assume that mutations of the human sequence tend to “undo” evolutionary improvements and cause mutated human profiles to regress towards chicken profiles. The three profile difference patches then agree well with the patterns of mutated aggregation rates [49].
The aggregation of globular proteins, such as well-studied lysozyme c (HEW), may involve unfolding and is thus more complex than that of Aβ, a known product of A4 fragmentation. The smallest lysozyme amyloid nucleus is 55-63 (9 aa) GIFQINSRY, called K peptide [51,52]. K peptide is the strongest amyloid former of nine related small (<9 aa) peptides over a pH range from 2 to 9. Profiles for entire human lysozyme in Figure 4 show that the 9 aa K peptide nucleus is located at the center of centrosymmetric α-β-α lysozyme. The 69 aa wide central β region 57-103 is hydropathically level, so its β strands are nonamphiphilic [49].
Level set synchronization, the case of HPV vaccine
Synchronized motions of actin cell skeleton proteins guide cell surface and interface deformations, a protein realization of Stokes’ theorem that also involves criticality [53,54]. Most proteins have>300 aa and their functionality at the molecular level also involves large-scale conformational motions which are optimized by synchronization. The first example of molecular level set criticality is likely to come as a surprise, as it concerns a single amino acid mutation of a 505 amino acid protein, which alters the self-assembly rate of protein complexes of Human Papilloma Virus (HPV) cervical cancer vaccine by a factor of 103 [55].
The long road that led to cervical cancer vaccines began in 1976 when Harald zur Hausen published the Nobel hypothesis that HPV plays an important role in the cause of cervical cancer. HPV is a large capsid protein, but it was found that only the 505 aa L1 part was needed to make a good vaccine that conformationally selfassembled into morphologically correct Virus-like Particles (VLPs). L1 from HPV 16, taken from lesions that had not progressed to cancer, self-assembled 103 times faster than the HPV 16 L1P that researchers everywhere had been using; the old strain L1P had been isolated from a cancer, which differed from L1 by only a single amino acid mutation D202H [56]. The huge increase in self-assembly rate could well be due to conformational synchronization, but this is not easily quantified using Newtonian methods.
Given the lower bound of L=9 in and the remarkable properties of L1, its profile Ψ (aa,9) with the fractal scale was examined near the 202 mutation site [53]. The striking feature is the presence of two almost level L1 hydrophobic peaks in the region between 191 and 231, shown enlarged in Figure 5. The narrow peak α is centered near 202, the mutated site distinguishing L1 from L1P. The level condition is satisfied to within 1% by L1, but by only 5% by L1P and by two other singly mutated strains recently added to the PDB. Note that no mutations were found in the stabilizing broad peak. Note also the deep hydrophilic minimum near 215, which functions as a plastic hinge accelerating self-assembly.

Figure 5: Hydroprofile of L1 and several single amino acid mutants, using the fractal scale Moret and Zebende (MZ). 
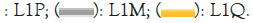
This example also brings out the advantages of scaling with Φ (aa, W). The excellent agreement shown in Figure 5 disappears when W is reduced to 7, below the cutoff in. It also disappears when Ψ MZ is replaced by Ψ KD. In other words, the pre-2000 efforts involved in constructing 127 different Ψ scales were exploring a good direction, but proteins are so complex that success with the MZ scale was possible only bioinformatically after the PDB structures became numerous and more accurate [56].
In addition to the types of HPV that cause cancer, there are “milder” types that cause only warts (self-limited growth). It might appear that the differences between these two types, which occur on a cellular level, could not be analyzed on a molecular level. However, there are many self-similar aspects to proteins and cells, so one should look at the differences between the HPV 16 (cancer) and HPV 6 (warts) profiles (Figure 6). There are large differences in the amide (N)-terminal region, far from the plasticity hinge (which is almost unchanged) seen in Figure 5. Thus the main self-assembly function is unchanged, but the N-terminal region changes can account for the sometimes serious side effects. These small differences are much reduced with the classic KD scale, which is suited to some large open-closed transitions, but not small conformational changes [56].

Figure 6: By aligning the extrema, the Moret and Zebende 9 (MZ 9) hydropathic profile reveals strong similarities (correlation coefficient (r) = 0.82) between L1 HPV16 (cervical cancer) and HPV6 (warts). The two large differences around 135 and 160 could be the major factor in the functional differences. 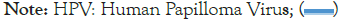
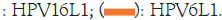
Level set synchronization, Coronavirus Darwinian evolution
Coronavirus 2019 has evolved to be much more dangerous than CoV2003. It differs greatly from other airborne viruses (like flu) because its infections are much more likely to be fatal. It is also much more contagious, with a median 5-day asymptomatic incubation phase that can extend to 14 days. Coronaviruses are large, roughly spherical particles with unique surface projections called Spikes (S). The S protein is composed of S1 and S2 subunits; the S1 subunit forms the head of the spike and has the receptor binding domain, while the S2 subunit forms the stem which anchors the spike in the viral envelope. There are two natural questions, why has the coronavirus 2019 been so successful and what is its source? These questions can be discussed by comparing coronavirus 2019 to coronavirus 2003, which has been much studied.
There are ~300 mutations in the 1200 amino acid sequences of CoV19 from Cov03. According to BLAST site-by site (W=1) sequence comparisons, the largest and most striking difference between CoV19 (human) and CoV03, as well as coronaviruses of many species, is the insertion of a 4 amino acid PRRA sequence near the S1/S2 cleavage site, Figure 1 of [56]. Experiments showed that this insertion facilitates spike trigenic reassembly after cleavage and is presumably the main factor responsible for the high severity of CoV19 infections. What is the cause of the asymptomatic incubation phase? Can only a few of these 300 mutations, far from the two cleavage sites, also be contributing to the extremely strong viral interaction of CoV-2? The extreme effectiveness of CoV19 suggests self-organized criticality, but how do we identify these possibly critical distal and so far unidentified sites [57]?
In Figure 7, the four hydrophilic minima of CoV-2, labelled 1-3 and S2’, are nearly equal at 140, whereas the similar minima of CoV-1 range from 131 to 147, especially the very deep minimum of CoV-1 at 559. The new sequence PRRA in CoV-2, inserted at 681 in CoV-1, the cleavage interface (S1/S2), has an average MZ hydropathicity 108.5. This lowers Ψ(aa,35) to 140.9 at the 3 minimum, aligning it with ~140 minima 1 and 2. The three minima span ~250 amino acids sites far from the S1/S2 cleavage, which makes their water-driven synchronization for CoV-2, but not CoV-1, outside the range of most simulation or modeling methods. It turned out later that it is this range that is responsible for the abrupt end to the pandemic. The spikes are long and stick out into water, so they appear to be ideally suited to hydropathic thermodynamic scaling [58]. As usual, we choose W=35 to maximize the hydropathic shape differences between CoV-1 and CoV-2, as measured by a maximum in their variance ratio. The two cleavage sites S1/S2 and S2’ of CoV-1 have moved lower (hydrophilically, further outside) in CoV-2 (Figure 7), consistent with the very accurate MZ scale [2]. When a cleavage segment is further outside, there is more space for cleavage and reassembly, which will occur more rapidly. The insertion Pre-Removal Risk Assessment (PRRA) in CoV-2 was identified with BLAST (W=1) as unique to CoV-2, but with BLAST alone one cannot show that this change also made CoV-2 more contagious, resulting in the deadly pandemic [56].

Figure 7: The hydropathic profiles of CoV-1 and CoV-2 reveal a hidden symmetry when plotted using the Moret and Zebende (MZ) scale (second-order phase transitions). 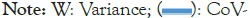
The hydropathic results shown in Figure 8 again exhibit level sets (synchronized extrema) for CoV-2 but not CoV-1, just as in many other protein profiles in this review (Figure 7). Viruses must act rapidly before being destroyed by antibodies and they could do this through synchronized motion by leveling their hydrophilic (outside) extrema. As shown in Figure 8, such a leveling of minima 1-3 occurs in CoV-2, while it is absent from CoV-1. The change in minimum 2 is especially striking; it is caused by a cluster of four critical mutations from CoV-1 to CoV-2. These synchronized minima provide a natural explanation for the occurrence of the asymptomatic incubation phase that has made CoV-2 so dangerous.

Figure 8: With the Moret and Zebende (MZ) scale panoramic spike profiles of CoV-1 and CoV-2 reveal a set of four central hydrophilic level extrema in CoV-2, but not in CoV-1. The new level set was identified with a cluster of four single mutations in 546-568. Note: W: 
The abstract predicted that, because critical synchronization is so easily disrupted, a “very successful” vaccine based on spikes was possible [59,60]. At this writing, this prediction appears to have been confirmed, by the report of spike-based vaccines that have been more than 90% successful in large scale trials. By comparison, flu vaccines usually achieve 30-50% success.
After CoV-2, natural selection of spike mutations was expected and these indeed occurred. Moreover, for several years (up to and beyond Omicron) only a few mutations made the level sets identified in CoV-2 steadily more level, confirming the validity of the level set mechanism [58,59]. Then the evolution of the spike changed directions; it made dynamics both more stable and more flexible topologically. This has not only increased contagiousness, but it also made the virus much less dangerous by suppressing trigenic fusion. This new direction continued for several very specific individual mutations closely tied to hydropathic extrema identified in CoV-2. The overall result was an abrupt end to the CoV pandemic [60-61].
Simple laboratory toy models of sub-critical self-organized in/ out globular shaping exhibit a variety of long-range interactions between surface droplets, analogous to protein amino acid very long-range interactions [62]. There is much interest in Hub proteins, which reside on the cytoplasmic side of the cell membrane, also described as a catalytic substrate supporting protein-protein interactions in the interfacial frontier space [63,64]. Profiles of some of these 8 proteins (lengths range from 304 aa to 976 aa) are interesting [65]. The connection between intrinsic disorder, conformational dynamics and Newtonian simulations has been reviewed (574 references) [66]. Nuclear Magnetic Resonance (NMR) experiments have shown that conformational dynamics in the catalytically relevant microsecond to millisecond timescale is optimized along a favored evolutionary trajectory in a bacterial protein [67]. Statistical methods alone applied to genome intron/ extron analysis failed to identify ordering of protein sequences [68]. This illustrates why the combination of topological, geometrical and structural data in scaling theory is needed to connect protein sequences to function and biomedical applications. An early example is the study of optimal folding [69].
The present methods, based on self-similarity near thermodynamic critical points, are accurate and rigorous modern realizations of multi-scale (also called coarse-grained) theory. In many cases coarse-graining occurs on a scale corresponding to W~3. It has become fashionable as a means of interpreting Newtonian all-atom molecular dynamics simulations [70]. Students will find a concise, self-contained introduction to many of the classical tools used here in [71]. A toy network model exhibiting self-organized criticality has been analyzed with Markov chains focused on self-stabilizing nodes, while similar results can be obtained simply from linear preferential attachment models. These models are analogous to the intermediate phase of rigidity percolation in network glasses [34,72- 74]. Criticality occurs in enzymatic networks through adaptive queueing [75]. There are many discussions of hydrodynamic flow of vortices between rotating cylinders, which are a simple liquid analogue of globular protein deformations, including hydropathic inside/outside effects with and without waves, for instance and a Wiki on Taylor-Couette flow (1923) [76,77].
Further historical notes: the classical discussion of short-range hydrophobic interactions is [78]. A “glassy” analysis of protein functionality shows solvent interactions [79]. Phase transitions may be used to organize spatially and biochemically regulate information throughout biology [80]. Young’s (1803) two-slit diffraction experiment proved the existence of light waves and is a standard part of introductory physics courses. Less well known, but also interesting, are Faraday’s (1831) standing waves [81-83]. These waves are classical and can be observed in a pail of water. Critical fluctuations have been analyzed by NMR, which presents conclusive proof that evolution is driving proteins toward critcically effective functionality [84]. Apart from potential biomedical applications, these thermodynamic results are consistent with activity-based enzyme profiling [85,86]. Many more examples of the methods discussed here can be found at arXiv [87].
In his enormously popular Dublin (1943) lectures and book, “What Is Life?”, Erwin Schrodinger proposed that we could progress in answering this question by using thermodynamics. A few years later, high energy physicists gave us hydrogen bombs, while solid state physicists gave us transistors and the structure of DNA. Over the next 50 years high energy physicists produced nuclear power and the LHC, while solid state physicists gave us solar power, the Internet and billions of fantastic gadgets. Molecular biologists joined molecular solid state physicists to generate an enormous data base of protein structures and functions, which have been recognized by >17 Nobel Prizes and formed a platform for miraculous medical treatments.
The historical protein path from Schrodinger’s classical discussion to modern thermodynamic scaling involves many modern technical tools from mathematics and theories of phase transitions. It also benefitted from intuitive insights and here special mention should be made of Walter Kauzmann’s work in the 1950’s. Kauzmann emphasized that proteins function reversibly on long time scales (ms) and behave like a deeply super cooled liquid with very high viscosity. Evolution has achieved Kauzmann’s network qualities by going beyond classical glassy “funnels” and approaching fractal critical points. Kauzmann anticipated the modern viewpoint through his emphasis on the key role played by hydropathic forces in shaping protein globules. Thermodynamic scaling may enable us to realize Schrodinger’s dream and advance new medical platforms where earlier work has stalled. Specifically, it exploits the modern protein database to describe the connections between amino acid sequences and protein functions with the accuracy of Schrodinger’s dream and the intuitive insights of Kauzmann. Proteins are the ultimate example of complexity, yet at the same time their evolution has been guided by the principles described here.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Phillips JC (2024) How Life Works: Darwinian Evolution of Proteins. J Proteomics Bioinform. 17:664.
Received: 03-May-2024, Manuscript No. JPB-24-30834; Editor assigned: 06-May-2024, Pre QC No. JPB-24-30834 (PQ); Reviewed: 20-May-2024, QC No. JPB-24-30834; Revised: 27-May-2024, Manuscript No. JPB-24-30834 (R); Published: 03-Jun-2024 , DOI: 10.35248/0974-276X.24.17.664
Copyright: © 2024 Phillips JC. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.