Medicinal & Aromatic Plants
Open Access
ISSN: 2167-0412
ISSN: 2167-0412
Research Article - (2016) Volume 5, Issue 3
Sutherlandia frutescens is a popular South African plant commercially available in a range of formulations. However, reference standards for quality and stability assessment are lacking. This work reports the development and validation of a reversed phase HPLC method for the analysis of flavonoid glycosides and their corresponding aglycones in S. frutescens products. Five materials containing either leaf powder (LP) or spray-dried aqueous extract (SDAE) of S. frutescens were analysed for flavonoid content. A primary objective was to isolate non-commercially available flavonoid glycoside compounds (sutherlandins) for use as reference standards. Sutherlandins A, B, C and D were successfully isolated, and used, with other flavonoid compounds for HPLC assay development. The developed HPLC method was linear in the range of 0.2 to 60 μg/ml for quercitrin; 0.2 to 120 μg/ml for quercetin and kaempferol; 0.2 to 200 μg/ml for rutin and kaempferol-3-O-rutinoside; 4 to 180 μg/ml for sutherlandins A and D; and 4 to 200 μg/ml for sutherlandins B and C. Percentage content of sutherlandins A, B, C and D, quercetin and kaempferol in different plant materials were significantly different (P<0.001). The developed HPLC method is simple, precise and robust; and can be employed for the simultaneous determination of flavonoid glycosides and aglycones for quality control of S. frutescens products.
Keywords: Flavonoid; HPLC assay; Sutherlandin; Quality control; Validation
Sutherlandia frutescens L., a popular plant of the family Fabaceae (pea and bean family or Leguminosae), is widely used in certain parts of Southern Africa for the treatment of various ailments [1]. Randomized placebo-controlled clinical trials have been conducted to assess its safety and efficacy in HIV patients [2,3] with preparations underway for another upcoming clinical study. While a variety of formulations from this plant are commercially available and thus can be used for the clinical trial, very little is known regarding their quality and stability profiles, as well as the content of actives in such formulations.
Flavonoid glycosides have been discovered and isolated from the plant as potential actives and possible quality control markers [4,5]. Much of the biological effects of herbal medicinal remedies have been attributed to the presence of flavonoids [6,7], which are known to exist in this plant in glycosidic and aglycone forms. These different forms have differing chemistries and, as such, can be said to be representative of the different forms and conditions under which S. frutescens actives occur in different environments. These forms are also susceptible to extreme modification and can be readily hydrolysed, oxidized, hydroxylated, methylated, glycosylated, acylated or phenylated, giving rise to the variety of compounds within a class [8,9]. As most forms of instability in medicinal products are due to oxidative and hydrolytic reactions, advantage can be taken of flavonoid susceptibility to modification, and this can be applied in the quality control of herbal drug formulations. As such, content of flavonoid glycosides and /or corresponding aglycones can be used as an indication of product quality or stability as the flavonoid glycosides may be converted to the aglycones under unstable conditions [10]. Avula et al. reported on the quantification of flavonoid and cycloartanol glycosides in commercial preparations of S. frutescens [11]. However, no method has been reported for the simultaneous determination of the flavonoid glycosides and their corresponding aglycones in S. frutescens products.
The aim of this study therefore was to develop and validate a simple HPLC method for the simultaneous estimation of nine flavonoid compounds (two flavonoid aglycones and seven corresponding flavonoid glycosides) in S. frutescens formulations, with a view to employing this in resource-constrained settings for quality control and stability studies of products derived from this plant. Four of the compounds (sutherlandins A, B, C and D) are not yet commercially available and had to be isolated from the plant material for assay development and validation. Other flavonoid compounds selected for the assay were rutin, quercitrin, kaempferol-3-O-rutinoside, quercetin and kaempferol.
Materials
Samples of S. frutescens leaf powder (LP, batch number: BN E16794), spray-dried aqueous extracts (SDAE 1, batch number: Ferl- DST/001-1210; SDAE 2, batch number: 1674; SDAE 3, batch number: 62265 and SDAE 4, batch number: 63067) were received from suppliers within South Africa. Identification of the source plant material was by a consultant botanist of the supplying company, and a voucher specimen (PSM 602) is deposited in Parceval Laboratories, Paarl, South Africa. Commercially available reference compounds – rutin was purchased from Sigma Aldrich; quercetin, quercitrin, kaempferol-3-O-rutinoside and kaempferol – were purchased from Extrasynthese (Genay Cedex, France). Their corresponding glycosides were isolated from the butanol fraction of an S. frutescens aqueous extract. Acetonitrile (HPLC grade) and formic acid (reagent grade) were from Sigma Aldrich and Merck respectively. All water used was obtained from an in-house Milli-Q purification system (Millipore, Milford, MA).
Instrumentation and chromatographic conditions: The HPLC system for the acquisition of chromatograms and UV spectra was an Agilent 1200 series HPLC system, equipped with an in-line degassing system (G1322A, Japan), quaternary pump (G1311A, Germany), auto loading sampler (G1329A, Germany), thermostatted column compartment (G1316A, Germany) and photo diode array detector (G1315B, Germany). Chromatographic separation was obtained using a Phenomenex Luna® C18 column (25 cm × 4.6 mm, 5 µm i.d.) with a compatible guard column, both maintained at 45°C. The mobile phase, consisting of water (0.01% formic acid) (A) and acetonitrile (0.01% formic acid) (B), was filtered through a 0.45 µm filter and degassed prior to use. The flow rate of the mobile phase was maintained at 0.8 ml/ min, injection volume was 20 µL, and peaks were separated according to the following linear gradient elution: 0 to 1 min (A:B, 82%:18%); 1 to 15 min (A:B, 82%:18% to 75%:25%); 15 to 20 min (A:B, 75%:25% to 65%:35%); 20 to 25 min (A:B, 65%:35% to 40%:60%); 25 to 26 min (A:B, 40%:60% to 82%:18%); followed by an equilibration with A:B, 82%:18% from 26 to 35 minutes. The eluent was monitored at several wavelengths over a range from 250 to 380 nm, the specific wavelength of interest being 370 nm. Data acquisition and processing was carried out using the OpenLAB™ CDS ChemStation Edition software (Agilent Technologies, Palo Alto, CA, USA).
LC-MS and LC-MS/MS analysis was conducted on a Waters Synapt G2 quadrupole time-of-flight mass spectrometer, connected to a Waters Acquity ultra-performance liquid chromatography (UPLC) and photo diode array (PDA) detector, using a method previously described by Albrecht et al. [12].
Methods
Isolation of reference compounds: Sutherlandins A to D were isolated from the n-butanol soluble portion of the aqueous extract of S. frutescens spray-dried aqueous extract (SDAE) (batch number: Ferl- DST/001-1210). The crude butanol extract was subjected to reversed phase column chromatography using C18 Strata™ solid phase extraction (SPE) cartridges for the isolation of flavonoid fractions. Conditioning of the SPE cartridge was with 100% acetonitrile followed by water (0.01% formic acid). The butanol fraction (dissolved in 50% acetonitrile: water) was applied uniformly to the SPE cartridges and the column eluted using a five step gradient elution with water: acetonitrile mixtures (0.01% formic acid) of decreasing polarity (0 to 100% acetonitrile). Six major fractions were obtained; fraction 3 was found to contain the sutherlandins, and was further fractionated using HPLC. The identity of the isolated compounds was confirmed by LC-MS/MS and NMR analyses.
Preparation of calibration standards: Stock solutions of the calibration standards (rutin, quercetin, quercitrin, kaempferol, kaempferol-3-O-rutinoside, sutherlandins A to D) were prepared separately by dissolving each standard in 50% aqueous methanol solution. Dilutions were made from these stock solutions to achieve concentrations from 0.2 to 60 µg/ml for quercitrin, 0.2 to 120 µg/ml for quercetin and kaempferol, 0.2 to 200 µg/ml for rutin and kaempferol-3- O-rutinoside, 4-180 µg/ml for sutherlandins A and D, and 4 to 200 µg/ ml for sutherlandins B and C. The calibration standards were assayed in triplicate and calibration lines constructed by linear regression of plots of peak area against concentration. The prepared standards were used to validate the developed HPLC method for linearity and range, precision, limits of detection and quantitation, and ruggedness / robustness [13,14].
Sample preparation for quantification of reference compounds in plant materials: Five samples of S. frutescens formulations, one or more of which may be utilised in a planned randomised controlled trial, were prepared using a modification of the method previously utilized by Avula et al. [11] as follows. Weighed sample of each material: LP or SDAE material was vortexed in 2 ml of 50% aqueous methanol, and sonicated for 30 minutes. This was followed by centrifugation at 3500 rpm for 15 minutes. The procedure was repeated thrice, and respective supernatants combined. The final volume was adjusted to 8 ml with 50% aqueous methanol and mixed thoroughly. Prior to injection, appropriate dilutions were made, and the solution filtered using a 0.45 µm nylon membrane filter. The first 1.0 ml was discarded and the remaining volume collected in an LC sample vial. Fifty µl of triplicate samples was injected for each plant material.
Identification of isolated reference compounds using liquid chromatography/mass spectrometry (LC-ESI-TOF)
In this study, non-commercially available flavonoid glycoside compounds: sutherlandins A, B, C and D were isolated from S. frutescens SDAE, with minimum percentage content of 98.9%, 89.7%, 99.6% and 98.6%, respectively, as determined from HPLC analysis. The minimum percentage values as reported by the manufacturers for the commercially available references – quercetin, kaempferol, rutin, quercitrin and kaempferol-3-O-rutinoside – are 99.0%, 90.0%, 94.0%, 98.5% and 98%, respectively.
The identity of the isolated reference compounds was confirmed by LC-MS/MS and NMR. The MS data is shown in Figure 1. Highresolution electrospray ionisation mass spectrometry (HR ESIMS) analysis provided the [M+H]+ molecular ions of the respective compounds in the positive ion mode. The protonated species [M+H]+, similar to those previously reported by Avula et al. [11], were therefore observed at m/z 741.1862, 741.1857, 725.1900 and 725.1913 amu, corresponding to the identities of sutherlandins A, B, C and D, respectively (Figure 1, Table 1).
| Reference compound | Exact mass (amu) | [M+H]+ (amu) | [M+H-sugar]+(amu) | Key fragment (amu) |
|---|---|---|---|---|
| Sutherlandin A | 740.1800 | 741.1862 | 609.1487 | 303.0484 |
| Sutherlandin B | 740.1800 | 741.1857 | 609.1448 | 303.0498 |
| Sutherlandin C | 724.1851 | 725.1900 | 593.1472 | 287.0545 |
| Sutherlandin D | 724.1851 | 725.9130 | 593.1486 | 287.0554 |
Table 1: LC-ESI-MS of isolated compounds identified using positive ion mode and key fragment analysis
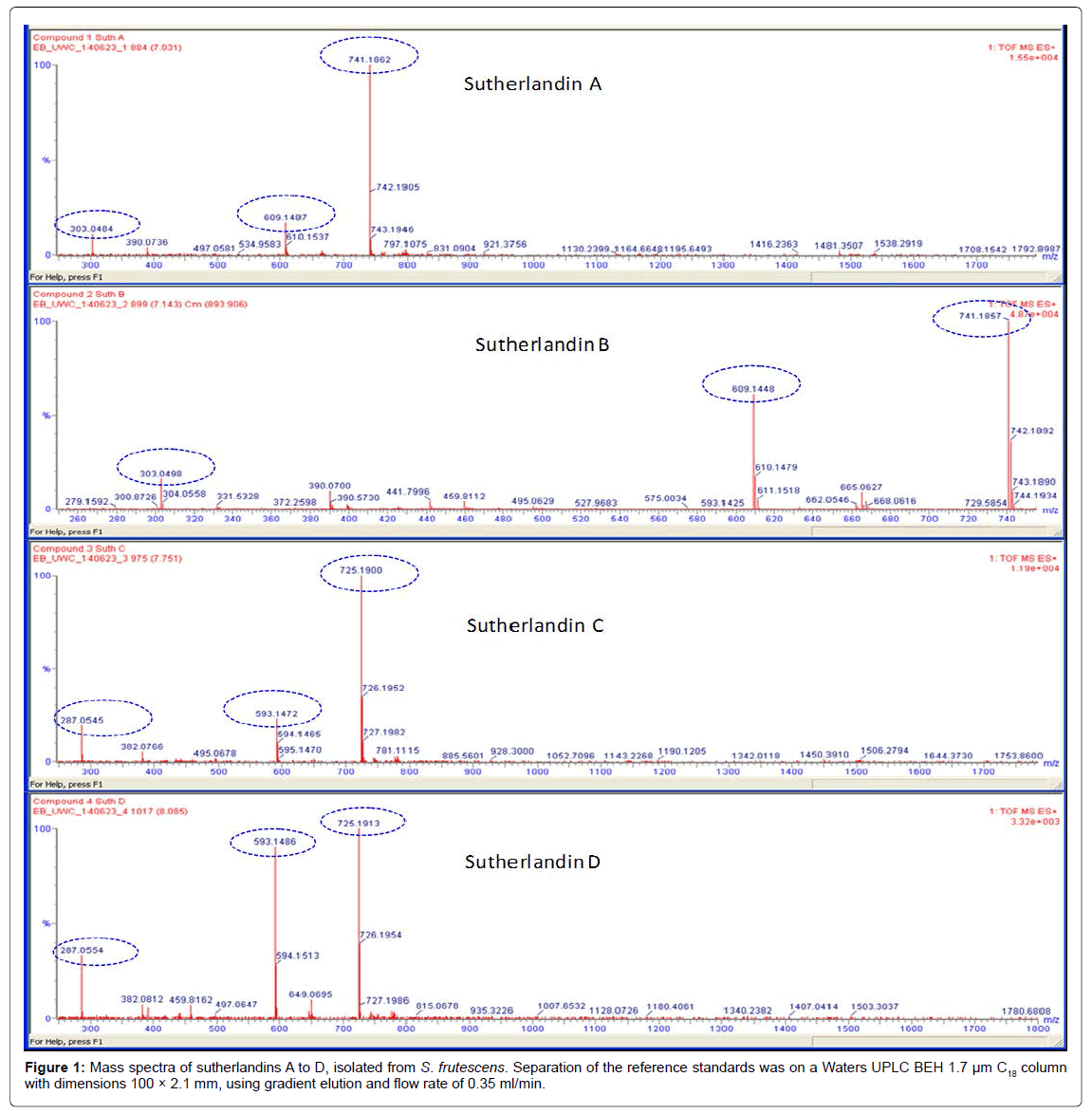
Figure 1: Mass spectra of sutherlandins A to D, isolated from S. frutescens. Separation of the reference standards was on a Waters UPLC BEH 1.7 µm C18 column with dimensions 100 × 2.1 mm, using gradient elution and flow rate of 0.35 ml/min.
Final confirmation of the identity of the isolated compounds was via NMR analyses, with results comparable to that previously reported [5].
A reversed phase HPLC separation method combined with PDA detection has been developed for flavonoid analysis in S. frutescens. The reference compounds were separated within 30 minutes by HPLC-DAD technique. A typical chromatogram from the assay is shown in Figure 2. The flavonoid glycosides eluted between 11 and 19 minutes, and the aglycones between 26 and 29 minutes.
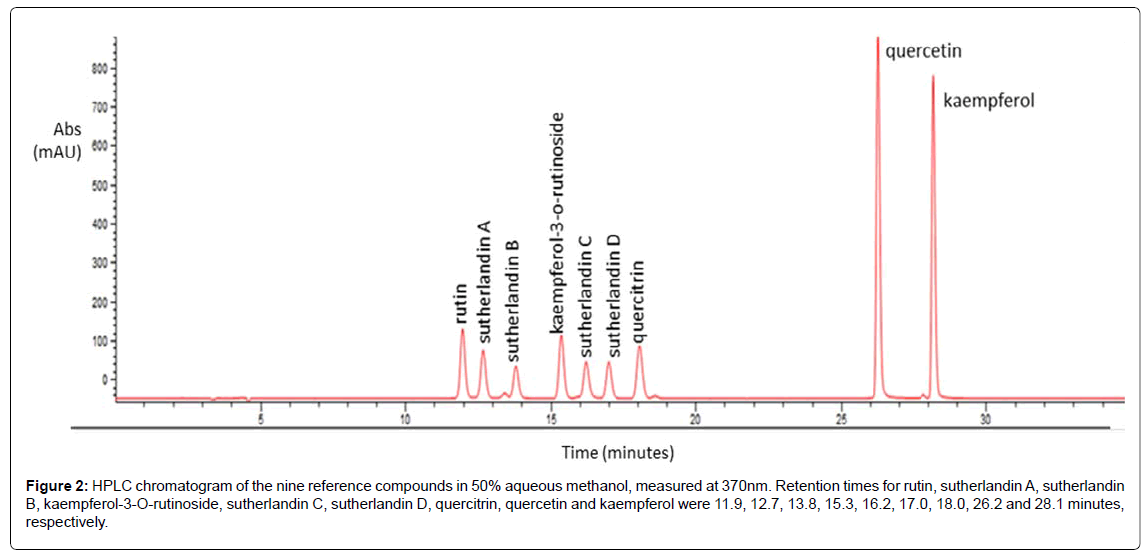
Figure 2: HPLC chromatogram of the nine reference compounds in 50% aqueous methanol, measured at 370nm. Retention times for rutin, sutherlandin A, sutherlandin B, kaempferol-3-O-rutinoside, sutherlandin C, sutherlandin D, quercitrin, quercetin and kaempferol were 11.9, 12.7, 13.8, 15.3, 16.2, 17.0, 18.0, 26.2 and 28.1 minutes, respectively.
Method validation
Calibration curves and linearity: The linearity for each of the reference compounds was assessed over the same concentration range as the calibration curves. The calibration curves for the nine reference standards showed a linear correlation between concentration and peak area (R2>0.99) of the detector response for the concentration ranges studied (Table 2).
| Analyte | Regression equation | Linearity range (µg/ml) | R2 | LOD (µg/ml) | LOQ (µg/ml) |
|---|---|---|---|---|---|
| Sutherlandin A | y=22.252x–9.5909 | 4.0 to 180.0 | 0.9998 | 1.0764 | 3.2617 |
| Sutherlandin B | y=14.543x+13.149 | 4.0 to 200.0 | 0.9996 | 2.2984 | 6.9649 |
| Sutherlandin C | y=17.890x+11.763 | 4.0 to 200.0 | 0.9990 | 0.3027 | 0.9173 |
| Sutherlandin D | y=17.450x–1.5755 | 4.0 to 180.0 | 0.9996 | 0.5110 | 1.5484 |
| Quercetin | y=108.85x+80.842 | 0.2 to 120.0 | 0.9970 | 0.1138 | 0.3447 |
| Kaempferol | y=92.167x+111.36 | 0.2 to 120.0 | 0.9983 | 0.3309 | 1.0026 |
| Rutin | y=27.929x+29.834 | 0.2 to 200.0 | 0.9989 | 0.8155 | 2.4712 |
| Quercitrin | y=27.381x–8.4190 | 0.2 to 60.0 | 0.9997 | 0.1163 | 0.3524 |
| K-3-O-rutinoside | y=26.799x+45.670 | 0.2 to 200.0 | 0.9991 | 0.0773 | 0.2343 |
Table 2: Regression equation, correlation coefficient (R2) and limits of detection (LOD) and quantitation (LOQ)for reference compounds by LC-DAD method.
Calibration curves and linearity: The linearity for each of the reference compounds was assessed over the same concentration range as the calibration curves. The calibration curves for the nine reference standards showed a linear correlation between concentration and peak area (R2>0.99) of the detector response for the concentration ranges studied (Table 2).
LOD=3.3 × (SD/S)
LOD=3.3 × (SD/S)
Where, SD and S are the standard deviation of the ordinate intercept and the slope of the calibration curve, respectively. Values for the LOD and LOQ are presented in Table 2.
Stability: Samples of the reference compounds were subjected to a variety of conditions (acid/base hydrolysis, freeze/thaw cycles, ambient light and heat exposure) and injected into the HPLC system to assess the analyte in the presence of any degradants which may arise under these conditions.
Acid/base hydrolysis
The results of acid-base hydrolysis are presented in Figure 3. The original chromatogram of each unhydrolysed compound showed only one noticeable peak (in green) in the flavonoid glycoside region for all seven flavonoid glycosides and in the aglycone region for the aglycones. On hydrolysis, the flavonoid glycosides were hydrolysed to the corresponding flavonoid aglycones (in purple), identified by comparison of their retention times and UV spectra with those of reference flavonoid aglycone compounds. This confirmed that the flavonoid glycosides are all O-glycosides, and thus prone to acid hydrolysis [15,16]. They can thus serve as references for quality control as their reactions under acid-base conditions can indicate instability.
As expected, the flavonoid aglycones were not affected by acid hydrolysis. Alkaline hydrolysis of the flavonoid glycosides led to the production of the deacylated flavonoid glycosides (navy traces, Figure 3).
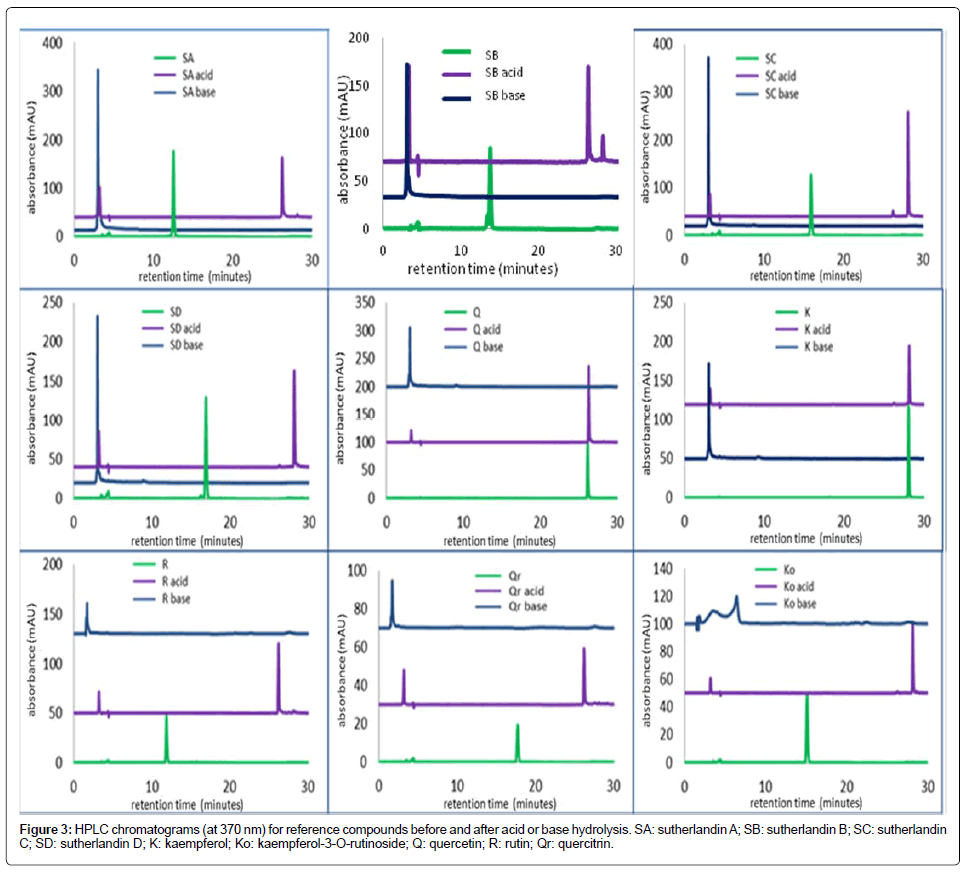
Figure 3: HPLC chromatograms (at 370 nm) for reference compounds before and after acid or base hydrolysis. SA: sutherlandin A; SB: sutherlandin B; SC: sutherlandin C; SD: sutherlandin D; K: kaempferol; Ko: kaempferol-3-O-rutinoside; Q: quercetin; R: rutin; Qr: quercitrin.
Freeze-thaw cycle
The results of the freeze-thaw cycle after five consecutive freezing (at –80°C) and thawing exercises (at room temperature), are presented in Figure 4. The reference compounds showed relative stability to five successive freezing and thawing cycles, as signified by the retention times and peak sizes before and after the freeze-thaw cycle. An exception was quercetin, exhibiting instability, shown by decrease in peak size, and formation of other products. This was in contrast to previous studies that demonstrated quercetin stability to freeze-thaw cycles, though the said studies evaluated quercetin stability in biological fluids [17,18]. Since this study utilized plant samples containing mostly flavonoid glycosides, the samples can be stored for prolonged periods in the freezer. This however may not be ideal if quercetin is to be used as a reference as it is prone to instability under freeze-thaw conditions as investigated herein, and may also be formed in cases of product instability.
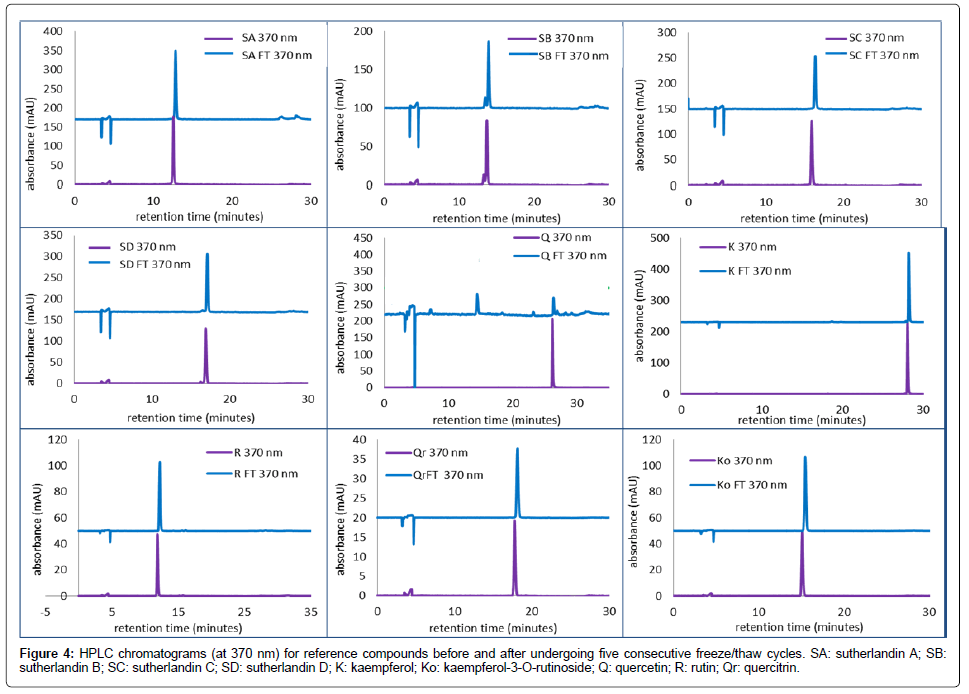
Figure 4: HPLC chromatograms (at 370 nm) for reference compounds before and after undergoing five consecutive freeze/thaw cycles. SA: sutherlandin A; SB: sutherlandin B; SC: sutherlandin C; SD: sutherlandin D; K: kaempferol; Ko: kaempferol-3-O-rutinoside; Q: quercetin; R: rutin; Qr: quercitrin.
Ambient light exposure
HPLC chromatograms of samples subjected to sunlight exposure for one month (Figure 5) indicates that the flavonoid glycosides (sutherlandins A, B, C and D) showed relatively good stability with respect to the retention times (i.e., still eluted at previous retention times) while the aglycones were not stable, with additional peaks, possibly from degraded products, arising in the chromatograms for quercetin and kaempferol (Figure 5). Retention times for the flavonoid glycosides were not affected by exposure to ambient light; however, the peak response for all the seven flavonoid glycosides decreased to between 45 and 97% of the original.
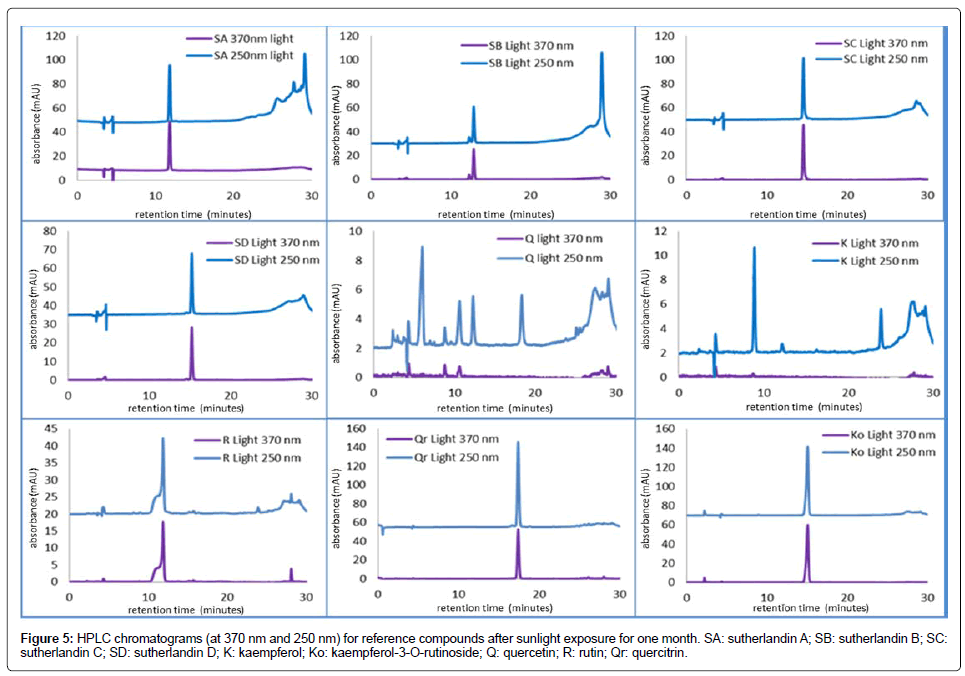
Figure 5: HPLC chromatograms (at 370 nm and 250 nm) for reference compounds after sunlight exposure for one month. SA: sutherlandin A; SB: sutherlandin B; SC: sutherlandin C; SD: sutherlandin D; K: kaempferol; Ko: kaempferol-3-O-rutinoside; Q: quercetin; R: rutin; Qr: quercitrin.
The peaks for the flavonoid aglycones, quercetin and kaempferol, were however not detected in the chromatograms following light exposure, perhaps due to the photo-oxidation of these compounds [19]. The stability of flavonoids to light is known to depend on the nature of the hydroxyl group attached to C3 of the flavonoid structure, where the absence or glycosylation of the hydroxyl group confers some degree of stability not seen in flavonoid molecules with a hydroxyl group at C3[20] as is the case with quercetin and kaempferol. The seven flavonoid glycosides under study are all glycosylated at position C3, conferring on them some degree of photostability not obtained with the non-glycosylated flavonoid aglycones, quercetin and kaempferol. This may explain why the flavonoid glycosides, though affected by ambient light exposure which served to reduce the peak response, were affected to a lesser degree than the flavonoid aglycones of lower photostability due to the presence of a free hydroxyl group at C3. The ‘lower-stability’ flavonoid aglycones underwent degradation to such an extent that their peaks were not detected in the chromatograms obtained following ambient light exposure. The lower phtostability of the flavonoid aglycones is ascribed to a greater triplet state population cum higher singlet oxygen reactivity [20].
All the compounds exhibited instability on exposure to light; ideally, samples containing these compounds should not be exposed to ambient light conditions as tested here. This also shows that the flavonoid compounds are susceptible to light and can therefore be used as reference compounds to monitor quality and stability of S. frutescensproducts exposed to light.
Heat exposure
Following heat exposure (for 3 hours at 60 degrees Celsius), all the flavonoid glycosides and aglycones were still detected at their respective retention times (Figure 6) indicating that heat did not cause exhaustive degradation of the compounds.
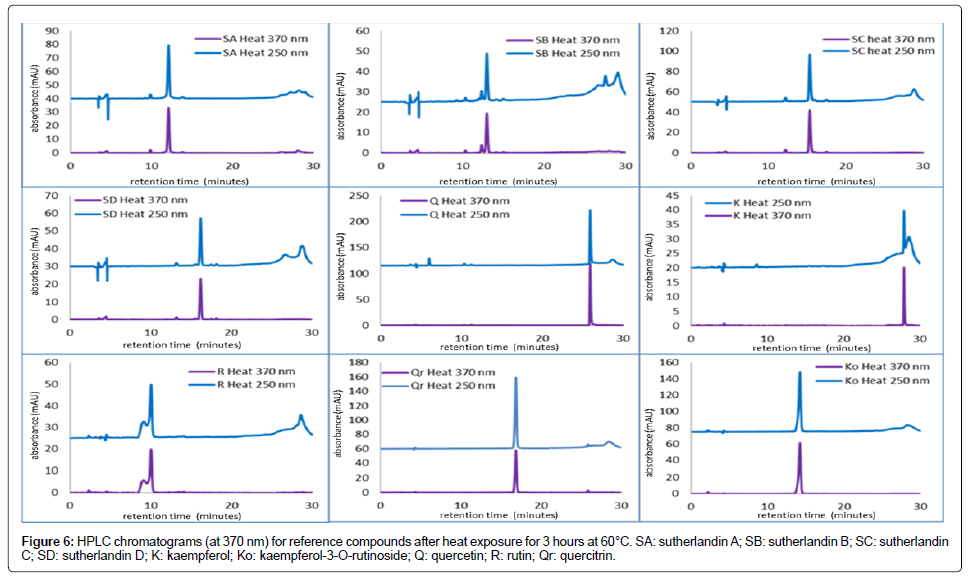
Figure 6: HPLC chromatograms (at 370 nm) for reference compounds after heat exposure for 3 hours at 60°C. SA: sutherlandin A; SB: sutherlandin B; SC: sutherlandin C; SD: sutherlandin D; K: kaempferol; Ko: kaempferol-3-O-rutinoside; Q: quercetin; R: rutin; Qr: quercitrin.
The peak response was however reduced to between 4 and 84% of the original for the flavonoid glycosides and aglycones under study. This is in line with a study by Manach et al. [21] which reported a 60% loss of quercetin in apple juice stored for 9 months at 25°Celsius. The flavonoid glycoside, kaempferol-3-O-rutinoside, where peak response increased to 106.8% after heat exposure was an exception from the other flavonoids in the group. However, such increase may also be viewed as being due to some form of instability or an indication of compromised quality. Studies by Zoricet al. have reported increased degradation of flavonol glycosides with increase in temperature. Ideally, samples containing these compounds should not be exposed to heat conditions as tested here. The flavonoid compounds, having shown susceptibility to heat, can therefore be used as markers for quality and stability assessment of S. frutescensproducts exposed to heat.
Precision and accuracy
The precision of the analytical method was determined by assaying 6 samples all at a concentration of 60µg/ml; the highest concentration for the analyte with the lowest upper limit. Intra-day and inter-day precision were assessed by assay of six replicate samples on the same day, over three different days. The relative standard deviations (% RSD) were calculated for the standard samples at the concentrations assayed and are presented in Table 3.
| Reference | Ave peak area | % RSD | Intraday Ave |
Intraday % RSD |
Interday Ave |
Interday % RSD |
|---|---|---|---|---|---|---|
| Sutherlandin A | 1325.99 | 0.998 | 1341.60 | 0.061 | 1328.03 | 0.442 |
| Sutherlandin B | 916.62 | 0.442 | 933.47 | 0.469 | 909.18 | 0.818 |
| Sutherlandin C | 1167.74 | 0.111 | 1130.98 | 0.452 | 1136.91 | 0.629 |
| Sutherlandin D | 1085.46 | 0.205 | 1087.62 | 0.056 | 1087.89 | 0.959 |
| Quercetin | 7134.14 | 0.146 | 7063.00 | 0.433 | 6987.52 | 0.327 |
| Kaempferol | 5952.61 | 0.242 | 5967.14 | 0.090 | 5989.19 | 0.494 |
| Rutin | 1836.46 | 0.738 | 1861.07 | 0.161 | 1853.79 | 0.355 |
| Quercitrin | 1646.19 | 0.045 | 1659.16 | 0.048 | 1662.54 | 0.457 |
| Kaempferol-3-o-rutinoside | 1759.65 | 0.019 | 1761.84 | 0.204 | 1782.92 | 0.502 |
Table 3: Intra- and inter-day precision analyses for reference compounds at 370 nm.
The results of intraday and interday assays were consistent, with% RSD less than 1% for all reference compounds (Table 3). The precision and accuracy data are therefore within acceptable limits for S. frutescensassay.
Robustness and ruggedness
Evaluation of method robustness was considered in mobile phase development. The robustness and ruggedness of the method were investigated by varying chromatographic parameters such as the flow rate and temperature. A decrease in the flow rate of the mobile phase (from 0.8 to 0.5 ml/minute) resulted in elongation of the retention times for all the reference compounds (Figure 7a), while changing the temperature of the column compartment from 40°C to room temperature did not cause significant changes in the peak sizes and retention times, showing that the developed method is robust to temperature changes (Figure 7b).
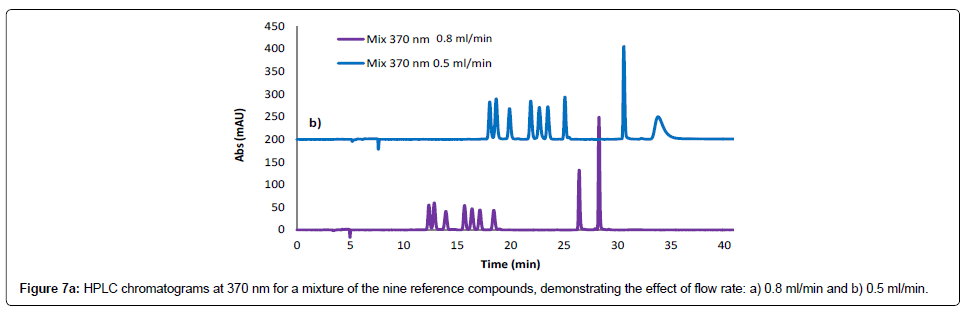
Figure 7a: HPLC chromatograms at 370 nm for a mixture of the nine reference compounds, demonstrating the effect of flow rate: a) 0.8 ml/min and b) 0.5 ml/min.
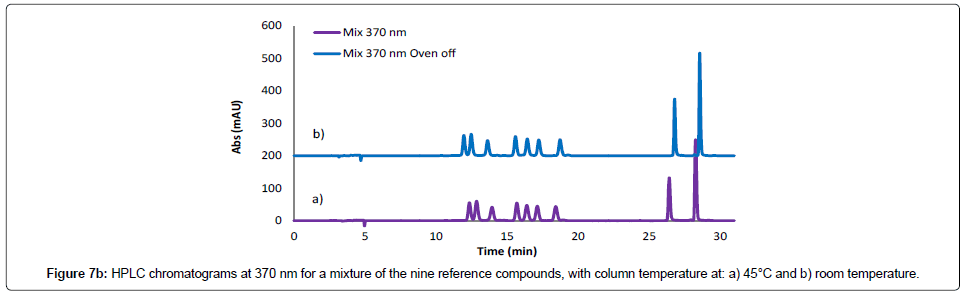
Figure 7b: HPLC chromatograms at 370 nm for a mixture of the nine reference compounds, with column temperature at: a) 45°C and b) room temperature.
System auitabilitytesting
System suitability parameters were calculated from the chromatograms obtained for each of the reference compounds during the study (Table 4). Values of the capacity factor (k*) ranged from 30 to 71, resolution (Rs) from 49 to 245 and tailing factor (T) from 1.0574 to 1.2563. The suitability of the chromatographic system in terms of column efficiency, resolution and precision, for flavonoid analysis in S. frutescensis therefore assured.
| Compound | k* | Rs | T |
|---|---|---|---|
| Sutherlandin A (SA) | 31.5870 | 81.5940 | 1.0714 |
| Sutherlandin B (SB) | 34.4686 | 83.9060 | 1.0574 |
| Sutherlandin C (SC) | 40.6013 | 61.4830 | 1.1421 |
| Sutherlandin D (SD) | 42.6056 | 48.9990 | 1.0843 |
| Quercetin (Q) | 66.3686 | 244.5770 | 1.1981 |
| Kaempferol (K) | 71.2636 | - | 1.2563 |
| Rutin (R) | 29.7370 | 85.0840 | 1.1117 |
| Kaempferol-3-O-rutinoside (Ko) | 38.3600 | 97.0670 | 1.1065 |
| Quercitrin (Qr) | 45.3070 | 97.9100 | 1.0903 |
Table 4: HPLC system suitability parameters for the referencecompounds.
Quantification of reference compounds in plant materials
The aim of this study was to develop a simple HPLC assay for the simultaneous determination of flavonoid glycosides and their corresponding aglycones in S. frutescensformulations.
Five S. frutescensmaterials (leaf powder, LP; and four spray-dried aqueous extracts, SDAE : SDAE 1, SDAE 2, SDAE 3 and SDAE 4) were analysed and their flavonoid glycoside and aglycone contents determined. The HPLC separation of reference compounds in the five S. frutescensmaterials analysed is presented in Figure 8. The flavonoid glycosides, sutherlandins A, B, C and D, represented by peaks 1, 2, 3 and 4, respectively, were detected in all the samples assayed. The flavonoid aglycones, quercetin and kaempferol, represented by peaks 5 and 6, respectively, were only detected in two of the SDAE materials and not in the LP material. Three of the nine compounds, rutin, quercitrin and kaempferol-3-O-rutinoside were not conclusively identified in the plant materials assayed. While peaks with similar retention times and UV-spectra as these three compounds were detected in the different S. frutescensmaterials, they were not isolated and so their identities could not be confirmed by further analytical techniques.
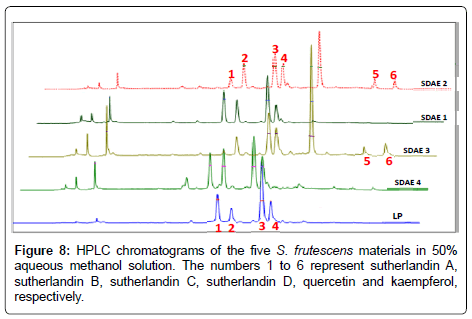
Figure 8: HPLC chromatograms of the five S. frutescens materials in 50% aqueous methanol solution. The numbers 1 to 6 represent sutherlandin A, sutherlandin B, sutherlandin C, sutherlandin D, quercetin and kaempferol, respectively.
The average peak area was used to quantify the reference compounds contained in each of the five S. frutescensmaterials. Values of the flavonoid content in the five S. frutescensmaterials, expressed as percentage content (w/w), and are shown in Table 5. The results show that of the two possible forms in which the flavonoids can exist in plant materials (glycoside and aglycone), the flavonoid glycosides were more abundant than the flavonoid aglycones in each of the assayedmaterials.
| Reference compound | LP | SDAE 1 | SDAE 2 | SDAE 3 | SDAE 4 |
|---|---|---|---|---|---|
| SA | 0.657 ± 0.006 | 0.724 ± 0.013 | 0.250 ± 0.003 | 0.808 ± 0.027 | 0.090 ± 0.002 |
| SB | 0.562 ± 0.042 | 0.746 ± 0.008 | 0.949 ± 0.006 | 1.255 ± 0.022 | 0.700 ± 0.002 |
| SC | 1.970 ± 0.011 | 1.506 ± 0.003 | 1.355 ± 0.001 | 1.678 ± 0.040 | 1.387 ± 0.017 |
| SD | 0.642 ± 0.022 | 0.553 ± 0.001 | 0.876 ± 0.023 | 0.972 ± 0.045 | 0.828 ± 0.025 |
| Q | ND | ND | 0.030 ± 0.000 | < LOQ | 0.018 ± 0.000 |
| K | ND | ND | 0.263 ±0.000 | < LOQ | 0.068 ± 0.000 |
Note: Each value is expressed as average percentage content (mg flavonoid x minimum purity of flavonoid reference/mg plant material) ± SD (n=3). Means within a row for each flavonoid compound are significantly different (p<0.001). Key: SA: sutherlandin A; SB: sutherlandin B; SC: sutherlandin C; sutherlandin D; Q: quercetin; K: kaempferol; ND: Not detected;
Table 5: Percentage (w/w) of reference standards found in the different S. frutescens extracts. Results are presented as percentage content (w/w).
The flavonoid aglycone content (0.02 to 0.21%) was found to be lower than that of the flavonoid glycosides (0.07 to 1.58%), and in some cases, below the quantification limit. The sprsy-dried extracts contained 0.02 to 0.26% of the flavonoid aglycones, detected in half of the SDAE investigated while these compounds were not detected in the LP samples. This suggests that the flavonoid aglycone content may be increased by processing parameters, such as is used in the preparation of the spary-dried extracts. However, no collaboration of this was found in existing literature.
In all the plant materials assayed, sutherlandin C was found to have the highest abundance, and so can be assayed for in all the S. frutescensmaterials. The least abundant flavonoid glycoside was not the same in all the plant samples assayed, with different materials showing non-uniformity in the flavonoid of least abundance. For instance, in the LP and SDAE 1 materials, the flavonoid glycosides of least abundance were sutherlandin B and sutherlandin D, respectively, while in the other three spray-dried materials, the flavonoid glycoside of least abundance was sutherlandin A.
Three out of the five materials did not contain detectable levels of the aglycones, quercetin and kaempferol. In the two spray-dried materials that contained the aglycones (SDAE 2 and SDAE 4), kaempferol content was more than quercetin content. The sum of the analysed flavonoids in the plant materials exceeds that obtained in other studies of total flavonoids [22-24], and suggests that the flavonoid content in the S. frutescensmaterials is sufficient for flavonoid use as reference marker compounds. Overall, the levels of the flavonoid glycosides and aglycones in the different S. frutescensmaterials assessed varied significantly (p<0.001). The assessment of flavonoid glycoside (sutherlandin A, B, C and D) content can therefore be used to differentiate between S. frutescensproducts.
The levels of the flavonoid aglycones (quercetin and kaempferol) in the analysed samples were quite low and in some cases, absent, thus making quantification of these compounds a challenge. The ratio of the flavonoid glycosides to the total flavonoids (glycoside + aglycone) was at least 90% in the samples studied. Therefore, the use of the flavonoid aglycones (and not the glycosides) as markers for quality control and stability assessment of S. frutescensLP or SDAE materials may not be justifiable or feasible usingthe specific assay employed in the present study. This however, can be expected in samples extracted using more polar solvents like methanol and water.It may well be that extraction with less polar solvents would yield more of the aglycones than the glycosides.
The simple, validated HPLC-DAD method reported was found to be appropriate for separation and quantification of flavonoid glycosides and their corresponding aglycones in different formulations of S. frutescens. The LP andSDAE materials all contained all four marker compounds, i.e.,sutherlandins A, B, C and D, with sutherlandin C being the most abundant in all the five materials. The two flavonoid aglycones, quercetin and kaempferol, were detected in two of the SDAE materials, and not in the LP material. There were more flavonoid glycosides than aglycones in all the plant materials, suggesting that the former may be more suitable as marker compounds. The use of the flavonoid glycosides as markers is therefore recommended for assessment of S. frutescensmaterials
The aglycones can also be used as marker compounds because their presence, as products of flavonoid glycoside breakdown, can indicate instability of the plant materials. However, use of the flavonoid aglycones levels alone, without the flavonoid glycosides levels as well, is not recommended. Assay of all six flavonoids employed here is preferred to the assay of a single flavonoid as it gives the profiles and levels of the flavoniods in the different materials. This in turn can indicate quality status of different S. frutescensproducts.
The authors acknowledge the Department of Science and Technology, South Africa, grant number DST/ICON 167/2010 dated 4 November 2010 and revised on 10 Oct 2013 for funding assistance. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare no competing interests.