Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2010) Volume 1, Issue 1
The accumulation of brominated fl ame retardants (BFRs) in the environment raises concern in light of observed detrimental effects on wildlife as well as on public health. We here present a recently modi fi ed method for the identi fi cation and quanti fi cation of the following selection of bromodiphenyl ether (BDE) fl ame retardants: BDE-17, -47, -66, -100, -153 and -183, in soil and sediments, using a new extraction procedure followed by gas chromatography mass spectrometry (GC-MS). Low- and high- resolution mass spectrometry (LRMS and HRMS, respectively) were compared and the latter was found to be superior with respect to both sensitivity and linear range. At LRMS mode the linear range was 3.8 – 19.2 ng/g dry weight (dw), while the use of HRMS more than doubled the linear range to 1.9 – 38.4 ng/g dry weight. Both methods were tested with regards to matrix associated effects on the limit of detection and quantitation. The use of HRMS yielded equal sensitivity for standards in solution and matrix. This was not the case when using LRMS. Here the limits of detection and quantitation were severely elevated by the matrix. Recoveries were comparable, but slightly higher at LRMS mode (77.0 – 121.9%) compared to HRMS (83.2 – 115.3%). The method described here is high throughput, low cost and will prove valuable in monitoring the levels of BFRs in the environment.
Keywords: GC-MS; Flame retardants; Soil; Sediment
The increasing levels of flame retardants in natural ecosystems are cause for concern due to their environmental impact and negative effects on public health. Polybrominated diphenyl ethers (PBDEs) are extensively used as flame retardants i.e. in plastics and textiles [1]. They easily leach into the surroundings and interfere with endocrine systems of humans and animals [2,3]. Due to their high affinity to lipophilic particles, they may persist for decades in the environment and have been found to accumulate to high concentrations in human surroundings [4]. Recently, the use of brominated flame retardants (BFRs) have become subject to strict legislation in many countries [5]. However, decades of widespread application have resulted in a disturbing increase in the concentration of BFRs in air [6], sediments [7,8], sewage sludge [9] as well as in fish, mussels, shellfish, birds and mammals [2]. The accumulation of BFR in the environment has spurred the development of a high number of analytical techniques for the detection and quantification of BFRs from a range of matrixes [10], but there is still no standardized method in existence. This underlines the need for an easy, fast and reproducible method for the control of flame retardants both in product monitoring and in health and environmental controls. We present an analytical method for the quantitation of a number of BDEs (BDE-17, -47, -66, -100, -153 and -183) in soils and sediments based on cyclohexane extraction followed by analysis by GC-MS. Sensitivity and linear ranges as well as matrix associated effects were compared at high and low resolution using a sector mass spectrometer with electric (E) magnet (B) Electric (E) geometry (EBE geometry) and electron ionisation (EI) conditions. The method developed in this study is high throughput and significantly less time consuming than the one presented by Wang et al. [11].
Chemicals and standards
The standard mixture used for identification and quantification (BDE-MXD, 5 µg/mL ± 5 % (1.2 mL dissolved in Nonane), Wellington Laboratories, Guelph, Canada) consisted of: 2,2’-dibromodiphenyl ether (BDE-17), 2,2’,4,4’-tetrabromodiphenyl ether (BDE-47), 2,3’,4,4’- tetrabromodiphenyl ether (BDE-66), 2,2’,4,4’,6-pentabromodiphenyl ether (BDE-100), 2,2’,4,4’,5,5’-hexabromodiphenyl ether (BDE-153) and 2,2’,3,4,4’,5’,6-heptabromodiphenyl ether (BDE-183).
Standard solutions were made by dilution of BDE-MXD, in cyclohexane to 9 standards ranging from 1.44 to 72 ng/mL. Decachlorobiphenyl (PCB-209), Supelco, Bellefonte, USA was chosen as an internal standard (IS) based on previous publications in the field [12,13]. A 100 µg/mL stock solution was prepared by dissolving 2 mg PCB-209 in 20 mL cyclohexane, and kept in the dark at -20°C. IS was added to the standard solutions at a concentration of 25 ng/mL.
Sample preparation
The matrix used for recovery studies was a Norwegian reference soil (loam with 36% sand, 45% silt, 19% clay, pH 5.5 and 2.5 organic carbon) at a test field at Kroer, Ås. Dry sediments from different sites in Europe where also investigated for occurrence of PBDEs. These samples were from the international sediment exchange for test on organic contaminants, SETOC. Dry soil and sediments (25 g) were weighed directly into Scott Duran bottles with an accuracy of two decimals. Milli-Q water (5 mL) was used to moisten the matrix. Acetone and cyclohexane (20 mL each; pro analysis Merck, Darmstadt, Germany) were chosen as extraction solvents. 5 µL of the IS was added to the extraction bottles. The samples were placed on a whirling table at 150 cycles/min for one hour before addition of 30 mL of milli-Q water and subsequent sentrifugation (2000 rpm, 5 minutes). This resulted in two phases: a lower water phase containing the acetone and an upper phase holding the cyclohexane and extracted organic compounds. 1 µL of the supernatant was injected into the GC-MS. The sample concentration was then calculated by internal standard calibration against the 9 point calibration curve which corresponds to a soil concentration ranging from 1.9 to 57.6 ng/g dw.
Instrumentation
The analysis was performed on a GC-MS system consisting of an Agilent 6890N GC and an Autospec Ultima mass spectrometer (Micromass Ltd, Manchester, UK), a three sector instrument with EBE geometry. Separations were performed on a Factor Four (5% phenyl, 95% dimethyl polysiloxan) capillary column (25 m x 0.25 mm I.D., 0.25 µm film thickness) from Varian (Middelburg, The Netherlands). The GC column was a low bleeding, slightly polar capillary column which was well adapted to congener specific determination of PBDEs. A methyl deactivated pre-column with ID 0.25 mm from Varian was connected by a pressfit connector (0.32 x 0.32 µm) from Teknolab, Norway. The capillary column was directly introduced into the ion source by an interface at 285°C. The pressure in the ion source was at 3.2 x 10-6 Pa and the temperature was 200°C. The initial temperature in the GC oven was 70°C for 3 minutes, followed by an increase of the temperature up to 150°C at a rate of 30% min. This was kept for 4.5 minutes. Further increase of the temperature to 325°C at 10% min was executed. After 5 minutes the oven temperature was programmed to reach a final temperature of 350°C at a rate of 70% min. Splitless injection of 1 µL of sample was performed with a CTC PAL Auto sampler from CTC Analytics AG, Switzerland. The temperature in the injector was 285°C and the liner was deactivated with dimethylchlorsilane (DMCS) and was of the type single taper. Helium (99.9999%, Yara AS, Rjukan, Norway) was used as carrier gas at a constant flow of 1.2 mL/min. The GC temperature program was optimized to achieve sufficient separation. The MS was operated in electron ionization (EI) mode. The optimal electron energy was found to be at 70 eV. This energy was also chosen by Wang et al. [11]. The mass range was from m/z 50 to m/z 800 in scan mode. The GC-MS data were collected using selected ion recording (SIR), selecting measured ions which had high intensity, high mass to charge ratio and were characteristic for the specific component. The software MassLynx 4.0 (SCN503) from Micromass was used to control the instrument and QuanLynx was used to quantify the components in the samples.
Quantification and lock mass
The relative response factor for all the PBDEs were set to be equal to the detector response for the IS, PCB-209. The results are based on peak area and all peaks were base line separated. The findings of flame retardants were verified by comparing the retention time and isotopic pattern in the spectrum of the found component with the retention time and isotopic pattern from a standard of the component, in addition to using the reference library NIST 05. For BDE-183, the molecular ion (M+, m/z 722) was chosen to be the SIRion. Fragment ions of the type M–Br2 + were present in the mass spectra of BDE-17, -47, -66, -100 and 153 and the isotope ions were subsequently used in the SIR experiments of these compounds. In the EI mass spectrum of PCB-209 (IS), the molecular ion was used as the quantification ion.
Determination of linear range
The linear range of the quantification was determined by making standard curves of the BDE standard mixture both in solution and in a soil matrix. A stock solution of BDE-MXD was made by diluting the standard solution with cyclohexane to a concentration of 240.0 ng/mL. Nine different concentrations of BDE-MXD where made by diluting the stock solution in cyclohexane. These standards were analyzed both before and after each sample series.
Estimation of the methods detection and quantification limits
Selected ion recording (SIR) were used to estimate LOD/LOQ in LRMS-mode. The LOD/LOQ ratio using HRMS was found by locating the highest intensity ion of each component. The SIR and quantification ions of each component are listed in Table 1. LOD and LOQ were estimated for two sets of standards in solution and in a soil matrix. These standards contained the PBDEs included in this study at concentrations of 1.44 ng/mL and 12.00 ng/mL, respectively, in cyclohexane (corresponding soil concentrations: 1.2 and 9.6 ng/g dw). The signal to noise ratio (S/N) was found for each component and LOD and LOQ were calculated. AS/N ratio of 3 gave the LOD, while a S/N ratio of 10 gave the LOQ. Peak area was used for quantification unless otherwise stated.
| PBDE | Quantification ion | Lock-mass ion | |||
| Ions for SIR* | Summary formula | Exact mass | Exact mass | ||
| BDE-17 | 245.9680, 247.9661, 248.9694 | C12H7BrO+ | 245.9680 | 202.9856 | |
| BDE-47 and BDE-66 | 323.8785, 325.8765, 327.8746 | C12H6Br2O | 325.8765 | 330.9792 | |
| BDE-100 | 401.7891, 403.7870, 405.7850, 407.7832 | C12H5Br3O | 403,7870 | 404.9760 | |
| BDE-153 | 481.6975, 483.6955, 485.6953 | C12H4Br4O | 483.6955 | 480.9696 | |
| BDE-183 | 719.4427, 721.4407, 723.4387, 725.4366 | C12H3Br7O | 721.4407 | 716.9569 | |
| PCB-209 (IS) | 493.6885, 495.6856, 497.6827, 499.6798, 501.6769 | C12Cl10 | 497.6827 | 492.9696 | |
*At LRMS all the SIR ions were used for quantification.
Table 1: The SIR ions, quantification ions and lock-mass ions for each component.
Determination of recovery and accuracy
Soil from Kroer in Ås, Norway was homogenized in a mortar to a fine powder. 75.0 g of the soil was transferred to a beaker and covered by acetone (1 cm above soil layer). PDBE stock solution (240 ng/mL) was added to a final soil concentration of 38.39 ng/g dw. The sample was homogenized by stirring the suspension with a glass rod and the acetone was allowed to evaporate under a fume hood until the soil was dry. Three 25.0 g portions were analysed according to the previously described procedure. The recovery of a given method is defined as the percentage deviation between the found results and the real values. Both LRMS and HRMS were used to determine the recovery percentage. Accuracy was determined by the standard deviations of recoveries between repeats. This was done to compare the accuracy of the two techniques of detection. The tests were performed on three different samples and they were also repeated with a one week interval.
All samples, both in solution and matrix were spiked with IS. PDBEs were extracted using acetone and cyclohexane (1:1) and the recovery, linear range, LOD and LOQ was determined by both LRMS and HRMS. A reconstructed ion chromatogram (RIC) chromatogram of a sample containing BDE-183, -135, -100, -47, -66 and -17 is shown in Figure 1.

Figure 1: A reconstructed ion chromatogram (RIC) chromatogram of a sample containing a) BDE-183, b) BDE-135, c) BDE-100, d) BDE-47, e) BDE-66 and f) BDE-17.
A method’s linearity is defined as the ability to obtain results directly proportional with the sample concentration [14]. In LRMSmode the results were linear in a concentration range corresponding to a soil concentration from 3.8 – 19.2 ng/g dw, while the results for the entire concentration range (1.9 - 57.5 ng/g dw soil concentration) related to a quadratic curve. The correlation coefficient was 99.93 – 99.99% for standards in solution and 99.53 – 99.83% for standards in soil matrix. The application of HRMS resulted in a much broader linear range (1.9 – 38.4 ng/g dw) and correlation coefficients comparable to LRMS (99.86 – 99.99% in solution and 98.81 – 99.93% for standards in matrix).
Determinations of LOD and LOQ were performed with standards in solution (1.44 and 12.0 mg/mL) and extracted from soil (1.2 and 9.6 ng/g dw) using both LRMS and HRMS. In LRMS mode the LOD and LOQ were calculated by using the TIC and a standard curve. When using HRMS the calculations were done using an internal standard with the highest intensity ion. In general, the values of LOD and LOQ in LRMS mode were higher when the standards were extracted from a soil matrix compared to solution. This indicates that the analytes may interfere with the matrix, which is not surprising in light of the heterogeneity, the physical and chemical complexity of soil. Matrix effects are expected to reduce in high resolution mode due to an improved selectivity. The results of LOD and LOQ at two different standard concentrations in solution are given in Figure 2a. The LOD of the different PBDEs varied from 4.2 – 25.9 ng/g dw (standard deviation (SD) 0.2 – 9.0) while the LOQ varied between 14 – 86.4 ng/g dw (SD 0.5 – 29.9). The standard with the concentration of 1.2 ng/g dw gave the lowest values of LOD and LOQ and SD. The LOD for BDE- 17, BDE-47 and BDE-100 was at or below 8 ng/g dw, for BDE-66 and BDE-153 12 ng/g dw and for BDE-183 19 ng/g dw. The results of LOD and LOQ for standards in matrix are each shown in Figure 3a. The detection limits of the different PBDEs varied from 12.4 – 62.6 ng/g dw (SD 1.1 – 7.6) while the quantification limits varied between 41.3 – 208.8 ng/g dw (SD 3.6 – 25.5). Except for BDE-100, the standard with the lowest concentration (1 ng/g dw) gave the lowest detection and quantification limits. The LOD for BDE-100 and BDE-153 was 17.6 ng/g dw or below in both standards. The other BDEs had significantly higher values of LOD.

Figure 2: The LOD 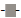 and LOQ
and LOQ 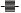 at a concentration of 1.44 ng/mL and the LOD
at a concentration of 1.44 ng/mL and the LOD 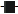 and LOQ
and LOQ 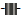 at a concentration of 12.0 ng/mL both in solution, using a) LRMS and b) HRMS.
at a concentration of 12.0 ng/mL both in solution, using a) LRMS and b) HRMS.

Figure 3: The LOD and LOQ at a concentration of 1.2 ng/mL and the LOD and LOQ at a concentration of 12.0 ng/mL both in soil matrix (corresponding soil concentrations: 1.2 and 9.6 ng/g dw), using a) LRMS and b) HRMS.
In contrast to the observations made in LRMS mode, we did not see any significant matrix associated effects on LOD or LOQ at HRMS mode. This lack of matrix effects on the results may be due to the innate higher selectivity of HRMS. LOD and LOQ at two different standard concentrations in solution are given in Figure 2b. The detection limits of the different PBDEs varied from 2.6 – 15.6 ng/g dw (SD 0.2 – 6.9) while the quantification limits varied between 8.5 – 52 ng/g dw (SD 0.5 – 23.0). The standard with the concentration of 1.2 ng/g dw gave the lowest values of LOD and LOQ. BDE-183 differed from the others by having approximately three times higher values of LOD and LOQ and a five times higher SD. LOD and LOQ for standards in a matrix are shown in Figure 3b. The detection limits of the different PBDEs varied from 2.3 – 14.6 ng/g dw (SD 0.2 – 7.9), while the quantification limits varied between 7.8 – 48.5 ng/g dw (SD 0.5 – 26.2). The component BDE-183 again differs from the other components by having three to six times the values of LOD and LOQ, in addition to a higher SD. A possible explanation for this may be decomposition of the component. In conclusion, using HRMS the values of LOD and LOQ as well as SD were generally lower compared to LRMS.
Table 2 gives a summary of the results for recovery and accuracy using LRMS and HRMS. In LRMS-mode the recovery average was 77.0 – 121.9% with an SD of 0.0 – 14.2. When using HRMS the recovery was 83.2 – 115.3%, SD 1.1 – 13. These values of recovery are significantly higher than those achieved by de la Cal et al. [12]. A recovery between 70 – 120 % is an acceptable result for PBDE [15].
| LRMS | HRMS | |||
| Average recovery [%] | SD* | Average recovery [%] | SD | |
| BDE-17 | 84.1 – 96.4 | 0.0 - 5.9 | 88.0 - 91.4 | 1.4 - 11.1 |
| BDE-47 | 82.8 – 110.5 | 0.4 - 6.6 | 84.4 - 92.4 | 1.7 - 7.7 |
| BDE-66 | 79.8 – 109.7 | 0.6 - 7.8 | 88.4 - 95.9 | 1.7 - 6.8 |
| BDE-100 | 77.0 – 110.8 | 0.6 - 6.1 | 83.2 - 96.6 | 1.1 - 6.2 |
| BDE-153 | 93.8 – 120.7 | 2.3 - 9.2 | 92.6 - 110.8 | 1.2 - 7.9 |
| BDE-183 | 84.3 – 121.9 | 0.5 - 14.2 | 90.0 - 115.3 | 3.8 - 13.0 |
*SD – Standard deviation
Table 2: A summary of the recovery of PBDEs using LRMS and HRMS.
Previously described methods for the extraction and quantification of polychlorinated biphenyl (PCB) have demonstrated the efficiency of acetone and hexane as solvents [16,17]. However, due to the relatively high volatility of hexane (bp 69°Cs) there is a risk of significant loss during sample preparation and thus analytical errors. Hexane is hazardous to human health [18] and it is essential to minimize exposure during handling. In comparison, cyclohexane is less volatile (bp 81°C) than hexane, reducing both the risk of exposure and sample loss. Therefore, replacing hexane with cyclohexane entails an improvement to the extraction method in several aspects. The method presented by Wang et al. [11] included a lengthy extraction using acetone and hexane followed by a large number of purification steps. While this yielded pure samples, high recoveries and resulting low LOD and LOQ, it was labour intensive and time consuming. The increasing levels of BFRs in the environment during the last decades calls for high throughput routine practices for the monitoring of these compounds. The modifications made by us enable fast extraction and analysis of a high number of samples from soils and sediments within a short time span and at low cost. The method is reproducible, has low LODs and LOQs and an acceptable recovery.