Journal of Glycobiology
Open Access
ISSN: 2168-958X
ISSN: 2168-958X
Review Article - (2014) Volume 3, Issue 1
Although originally considered merely structural components of cellular membranes, glycosphingolipids (GSL) are now recognized as having critical effects on cellular physiology, including proliferation, differentiation, viral transformation and ontogenesis. In addition, a vast majority of human cancers have modified GSL composition compared to parental normal cells. These modifications may contribute to both tumor survival and exert striking effects on anti-tumor immunity. In this review, we discuss mechanisms of immune modulation by tumor-secreted GSL.
<Keywords: Glycosphingolipids; Anti-tumor immunity; Cancer
Since their first isolation from human brain by JLW Thudichum in the late 1800’s, glycosphingolipids (GSL; lipid-rich molecules, widely expressed in normal tissues) were considered mere structural components of cell membranes. Over time, numerous reports have described changes in GSL composition and levels associated with cell proliferation, differentiation, viral transformation and ontogenesis [1].
A vast majority of human cancers exhibit striking differences in GSL composition when compared to parental normal cells (reviewed in [2]). These modifications may contribute to both tumor survival and exert striking effects on anti-tumor immunity. In this review, we describe mechanisms of GSL-mediated immune-modulation.
Glycosphingolipids (GSL) are a group of complex lipids that exhibit amphipathic features, allowing their insertion into cellular bilayers. Their amphipathic nature derives from the presence of a lipid region (consisting on N-acylsphingosine, or ceramide) covalently linked to a glycosidic region [3], as exemplified in Figure 1a. Ceramide may contain variations in either the sphingosine or the fatty acid (such as chain length, hydroxylation and/or saturation degree). Regarding the glycosidic region, it can vary in number and sequence of glycans, which in addition can have variability in their glucidic position and anomeric configuration. This structural versatility might explain the existence of more than 200 different GSL known up to date, which have been purified from a wide diversity of eukaryotic organisms [4]. This variability is relevant because, just as the primary sequence of proteins influences their secondary and tertiary structures, the specific oligosaccharides present in GSL, glycoproteins and other glycoconjugates affect the molecules’ three-dimensional conformation [5].
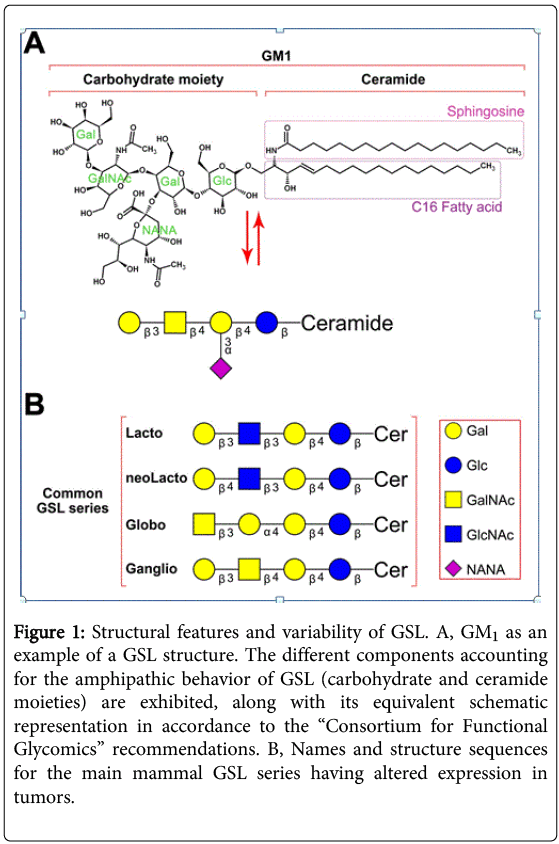
Figure 1: Structural features and variability of GSL. A, GM1 as an example of a GSL structure. The different components accounting for the amphipathic behavior of GSL (carbohydrate and ceramide moieties) are exhibited, along with its equivalent schematic representation in accordance to the “Consortium for Functional Glycomics” recommendations. B, Names and structure sequences for the main mammal GSL series having altered expression in tumors.
GSL are classified based on the closest neutral glycosidic sequences bound to the ceramide domain. Each group of GSL being metabolic intermediates along the different biosynthetic pathways that sequentially add carbohydrates to ceramide is known as a “series”. These neutral core structures can further elongate by adding a variable number of molecules, including N-acetyl neuraminic acid (sialic acid) residues. The major GSL correlating with specific tumors belong mostly to four series: lacto-, neolacto-, globo- and ganglio-series. Figure 1b exhibits the core sequences for these four main series.
GSL make up a significant portion of most eukaryotic cell membranes, adopting unique patterns for each cell type. Molar ratios of these molecules related to other major membrane lipids (such as phospholipids, cholesterol and glycerolipids) range from very low (i.e., <5% in erythrocytes) to relatively high (i.e., >30% of total lipids on oligodendrocytes plasma membrane, [6]). Surface-labeling experiments indicate GSL are inserted in the external layer of the cell through ceramide, and by their presence in the membrane regulate cell signaling. This is accomplished by altering the distribution of receptors in the cell membrane and by directly interacting or forming lipid domains that have preferential affinity for certain proteins, thus modifying their function [7]. GSL may also modulate cellular physiology by specifically binding other GSL or adjacent cell proteins to influence cell-cell or cell-environment interactions that regulate differentiation, cell growth and tissue organization [8-15].
General mechanisms of GSL synthesis. GSL are not direct products of genes. Instead, they are synthesized through a complex series of coordinated biosynthetic pathways comprised of several integral membrane glycosyltransferases. Eukaryotic GSL synthesis begins in the endoplasmic reticulum (ER) with the formation of the lipophilic ceramide tail. The β-addition of either a galactose (on the luminal face of the ER) or a glucose residue (on the cytoplasmic face of the ER and early Golgi complex) to the 1-hydroxyl of ceramide generates GalCer or GlcCer, respectively. Once at the Golgi complex, GalCer can be sulfated to produce acidic GSL (Sulfatide). Incorporation of galactose to GlcCer yields lactosylceramide (LacCer), a common precursor for the base structures of the different GSL series described in Figure 1.
Most LacCer are further elongated at the Golgi lumen by a series of glycosyltransferases that define the composition of GSL in a cell by their expression and intracellular distribution. These enzymes are regulated at the transcriptional, translational or post-translational level [16-18]. Additional regulation occurs through the availability of the sugar nucleotides and precursor acceptors as different enzymes may compete for the same substrate. Furthermore, different GSL glycosyltransferases may associate to form functional “multiglycosyltransferase” complexes that ensure progression to the preferred end product without releasing intermediate structures [19].
Changes in GSL synthesis in tumors. Aberrant GSL glycosylation occurs frequently in tumors (see Figure 2 [2]). The changes may be due to incomplete synthesis of certain GSL with the accumulation of precursors [20]. Early branch point alterations in the normal biosynthetic pathway can thus affect the amount of one class of GSL structure and allow the dominance of another to emerge. Epigenetic changes diminish expression of specific transferases (e.g. B4GalNAcT2, ST3Gal6, ST6GalNAc6 and GlcNAc6ST) in gastrointestinal tumors, altering the enzymatic activity of the concert of transferases and thus causing the accumulation of sialyl Lewisx and sialyl Lewisa [21,22]. In addition, hypoxic conditions can increase the expression of HIF (hypoxia inducible factor), which induces the transcription of glycosyltransferases FUT7 and ST3Gal-1 and transporter UGT1, also leading to sialyl Lewisx and sialyl Lewisa formation [23]. In adult T-cell leukemia, caused by Human T-cell Leukemia Virus type 1 (HTLV-1), the transcription of the FUT7 is constitutively activated by Tax protein (a viral molecule), increasing sialyl Lewisx synthesis [24].
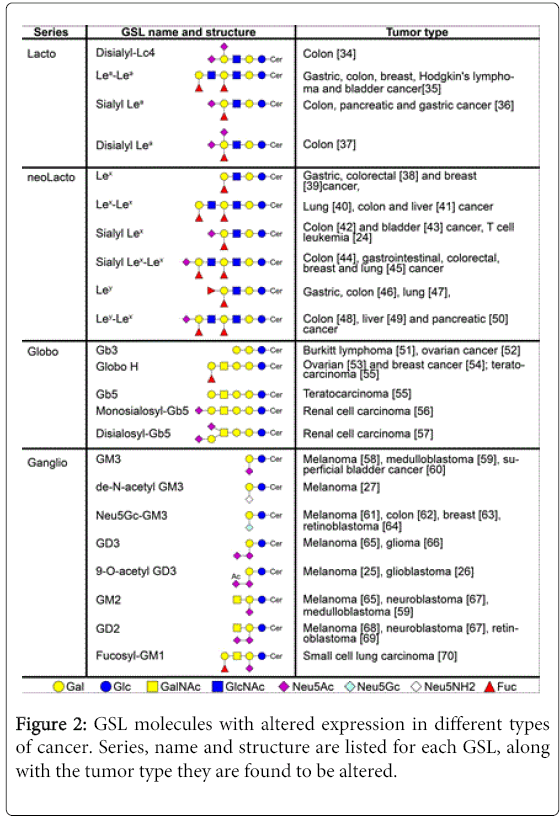
Figure 2: GSL molecules with altered expression in different types of cancer. Series, name and structure are listed for each GSL, along with the tumor type they are found to be altered.
Enhanced “neosynthesis” of GSL, which are minimal or absent in normal cells or tissues also leads to aberrant GSL in tumors [20]. Abnormal sialylation is a frequent alteration on tumor cells. For example, malignant melanomas have 9-O-acetylated GD3 instead of GD3 found in normal cells [25] and a critical ratio between GD3 and 9-O-acetyl GD3 on glioblastoma promotes tumor survival [26]. In fact, restoring GD3 by acetyl group cleavage reduces tumor cell viability by induction of mitochondrial-mediated apoptosis.
Another example of abnormal sialylation is represented by the loss of an acetyl group of GM3 (de-N-acetyl GM3) [27], leading to enhanced EGF-dependent tyrosine kinase activity of the epidermal growth factor (EGF) receptor while GM3 in normal cells inhibits EGF-stimulated tyrosine phosphorylation of the EGF receptor [28]. In addition, de-N-acetyl GM3 increases the expression and activation of urokinase-like plasminogen activator (uPA) and activates matrix metalloproteinase-2 in metastatic melanoma, thus stimulating cell migration and invasion [29].
Aberrant GSL glycosylation in tumors may also occur due to the increased use of recycling pathways. Although humans lack the enzyme to convert N-acetylneuraminic acid (Neu5Ac) to N-glycolylneuraminic acid [30], dietary uptake provides Neu5Gc through lysosomal sialidase activity on endocytosed Neu5Gc-bearing glycoconjugates. This free Neu5Gc is then transported into the cytosol, reactivated with cytidine triphosphate (CTP) in the nucleus [31], and preferentially utilized by tumors [32]. Thus, the usual Neu5Ac molecule is replaced by Neu5Gc, leading to the aberrant expression of Neu5Gc in tumors. This molecule, which differs from Neu5Ac by a single oxygen atom, has been shown to sustain low-grade chronic inflammation that can facilitate tumor progression through growth-promoting and angiogenic signals [33].
Increased levels of aberrant GSL are present in patients with various tumors. The idea that tumors might shed GSL into the extracellular space dates back to an observation nearly 40 years ago in breast cancer patients who had elevated serum levels of sialic acid-containing GSL [71]. Portoukalian et al. later found that the same GSL shed by melanomas were also enriched in erythrocyte plasma membranes of melanoma patients [72]. These associative findings suggested a clinical relevance for dysregulated GSL synthesis and shedding. In fact, increased levels of circulating GD2 in patients with neuroblastoma at the time of diagnosis were correlated with faster disease progression and lower survival rate [73]. In addition, GD3 levels in sera from patients with different stages of astrocytomas and glioblastomas were proportionally higher with more aggressive World Health Organization histologic grade [66]. Similarly, serum fucosyl-GM1 levels in small cell lung carcinoma patients positively correlated with the presence of multiple site metastases [70]. Lastly, serum levels of total gangliosides correlated with clinical course in melanoma patients after immunotherapy with a therapeutic cancer vaccine [74].
Figure 3 summarizes the process of shedding and uptake. The physiologic significance of GSL shedding and uptake is unknown. By transferring molecules, GSL shedding provides a mechanism to alter diverse surface properties on donor and/or acceptor cells [75-79]. In tumors, GSL shedding occurs rapidly (≈0.1 to 0.5% of total cell GSL per hour [59,67]). Although the concentration of GSL in the extracellular milieu depends on the GSL pattern of the tumor (as seen for human malignant melanomas [80]), shorter lengths of the ceramide fatty acyl chain may determine the preferential shedding of various GSL by different tumor cells [81,82] and also may influence their immunosuppressive activity.
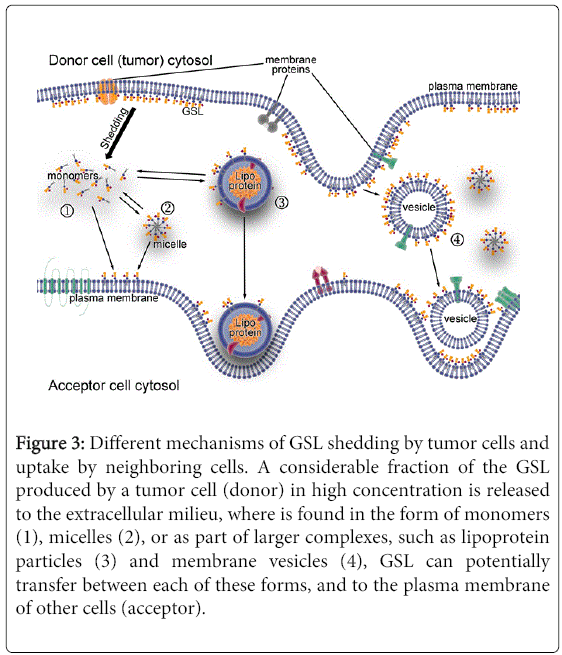
Figure 3: Different mechanisms of GSL shedding by tumor cells and uptake by neighboring cells. A considerable fraction of the GSL produced by a tumor cell (donor) in high concentration is released to the extracellular milieu, where is found in the form of monomers (1), micelles (2), or as part of larger complexes, such as lipoprotein particles (3) and membrane vesicles (4), GSL can potentially transfer between each of these forms, and to the plasma membrane of other cells (acceptor).
In neuroblastoma patients, circulating GD2 is largely bound to serum lipoproteins, approximately 75% binding LDL [83]. Dual chamber culture experiments have documented cell to cell transfer of GSL shed from lymphoma [84] and medulloblastoma [85] cell lines to human fibroblasts. Interestingly, inhibitors of GSL synthesis diminish the shedding and transfer of potentially biologically active GSL from the tumor [85], and also reduced the ability of murine melanomas to form tumors [86].
Studies on ovarian carcinoma cell lines suggest GSL-containing membrane vesicle shedding originates in plasma membrane domains enriched in GSL and caveolin-1 [87]. However, the shed GSL can be found in different forms that include not only large complexes (membrane vesicles), but also micelles and monomers [88]. Analyses on the relative composition of GSL suggest they could also be shed from cell membranes via a mechanism of preferential release for some particular GSL components [82].
The advent of immunotherapies for cancer has resulted in durable clinical responses and confirmed that immune responses may prevent cancer progression [89]. Moreover, tumor-infiltrating immune cells correlate with a positive outcome in many types of cancer [90-93]. However, chronic inflammation is associated with cancer, and certain immune cells (e.g., T regulatory cells) are associated with disease progression [94]. To resolve this paradoxical relationship between cancer and the immune system, a current paradigm is referred to as immune editing [95]. Immune elimination of tumor cells selects for tumor cells that are poorly recognized, which may ultimately escape immune recognition when the tumor creates an immunosuppressive environment. Tumors may shed different GSL to create conditions for evading immune surveillance, leading to a favorable environment for tumor progression. The GSL shed by the tumor may alter the membrane composition of infiltrating immune cells and inhibit their function, allowing tumor escape.
Monocytes, macrophages, and dendritic cells (DC). Cells of the innate immune system, including monocytes, macrophages, and dendritic cells, control tumors by killing tumor cells directly, secreting inflammatory mediators that recruit and differentiate adaptive immune cells, and presenting tumor antigens to T cells [96-98]. However, the latter two (indirect) antitumor functions of innate immune cells are inhibited by GSL in general, and particularly by those altered GSL found on tumor cells (see Figure 4, parts “1” and “2”).
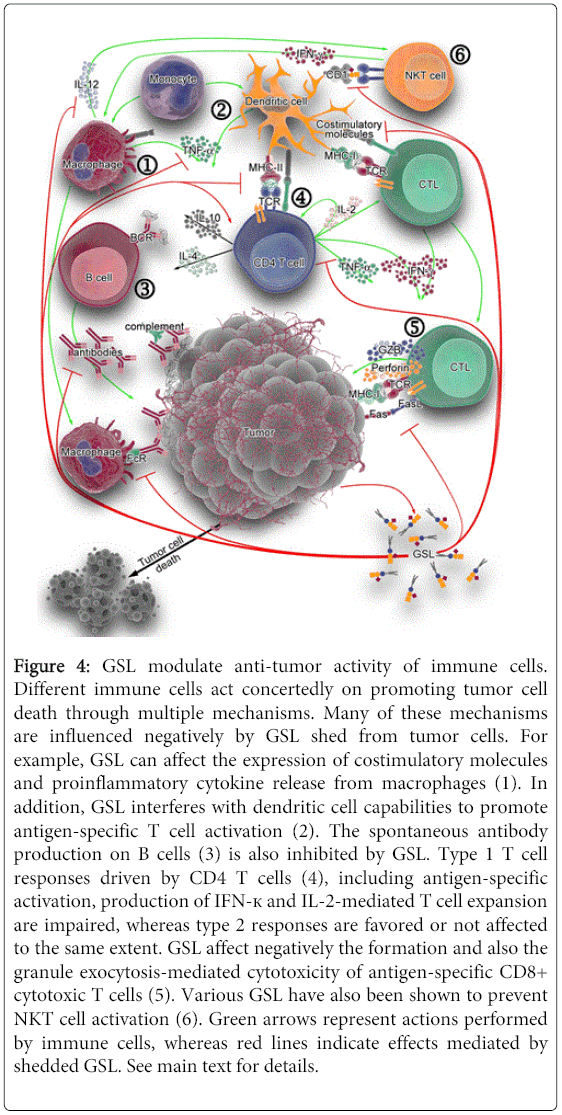
Figure 4: GSL modulate anti-tumor activity of immune cells. Different immune cells act concertedly on promoting tumor cell death through multiple mechanisms. Many of these mechanisms are influenced negatively by GSL shed from tumor cells. For example, GSL can affect the expression of costimulatory molecules and proinflammatory cytokine release from macrophages (1). In addition, GSL interferes with dendritic cell capabilities to promote antigen-specific T cell activation (2). The spontaneous antibody production on B cells (3) is also inhibited by GSL. Type 1 T cell responses driven by CD4 T cells (4), including antigen-specific activation, production of IFN-к and IL-2-mediated T cell expansion are impaired, whereas type 2 responses are favored or not affected to the same extent. GSL affect negatively the formation and also the granule exocytosis-mediated cytotoxicity of antigen-specific CD8+ cytotoxic T cells (5). Various GSL have also been shown to prevent NKT cell activation (6). Green arrows represent actions performed by immune cells, whereas red lines indicate effects mediated by shedded GSL. See main text for details.
Monocyte/macrophages. Monocytes exposed to bovine brain gangliosides [99], human brain gangliosides [100], or purified GD1a [101] fail to stimulate antigen-specific lymphoproliferative responses. Gangliosides prevent up-regulation of the co-stimulatory molecule CD80, whereas expression of ICAM-1, LFA-3, HLA-DR, and CD86 is not affected. Accordingly, monocytes pre-incubated with GD1a also show inhibition of CD80 up-regulation, down-regulation of CD40 and reduced IL-12 and TNF-α release in response to LPS stimulation [101], events that could be mediated by impeding NF-κB mobilization [102]. Moreover, previous exposure of monocytes to GM2 and GM3, or to GM1 and GD3, exhibit reduced Fc receptor expression and IL-1 production, respectively [103]. GM3, in particular, inhibits the VEGF-induced in vivo and in vitro expression of adhesion molecules (ICAM-1 and VCAM-1) on endothelium (in a mechanism mediated by activation of NF-κB via AKT signaling), and reduces monocyte adhesion to endothelial cells [104]. All these events lead to impaired leukocyte recruitment to endothelial cells under inflammatory stimuli, ultimately resulting detrimental to an effective anti-tumor immune response.
Dendritic cells. Development and function of monocyte-derived DC can be affected by gangliosides including GM2 [105], GD1a [106], GM3 and GD3 [107] in vitro, suggesting GSL may influence DC differentiation in vivo. The presence of GM2 changes the morphology, adherence, and cell surface receptor expression of DC precursors. For example, MHC class II molecules, costimulatory molecules and CD116 (GM-CSF receptor) are significantly reduced, as well as their endocytic, chemotactic and T cell proliferation-inducing activities. DC preincubated with GD1a poorly up-regulate costimulatory molecules and exhibit reduced IL-12 and increased IL-10 production when exposed to antigenic bacterial toxins [106]. Meanwhile, GM3 and GD3 strongly down-regulate CD1a, CD54, CD80 and CD40 molecules, impair allostimulatory functions, reduce IL-12 and increase IL-10 production, and induce caspase 3-mediated apoptosis [107]. The mechanisms by which GM3 and GD3 induce apoptosis differ in that GM3 causes accumulation of sphingomyelinase-generated ceramides and GD3-induced apoptosis is dependent on reactive oxygen species [108]. Although mature DC appear to be resistant to the GM2-mediated inhibitory effect, GD1a-preincubated DC exhibit no CD80 up-regulation, and also downregulate CD40, IL-12, TNF-α and IL-6 in response to LPS [101]. These activated DC show evidence of NF-κB activation disruption, which may account for ganglioside interference with APC expression of costimulatory molecules and cytokine secretion. In summary, GSL dysregulation in tumors acts to prevent T cell recognition by interfering with various macrophage and DC functions.
B cells. B cells in the tumor environment may produce antitumor antibodies that target tumor for killing by effector cells, as well as present antigens to T cells. In vitro studies show that GM2 ganglioside inhibits spontaneous immunoglobulin production on isolated B cells, by inhibiting endogenous IL-10 and TNF-α production [109]. GD1b or GT1b also can affect B cells by inhibiting spontaneous IgG, IgM and IgA production; however the effect is indirect in that treatment of T cells or isolated monocytes with GD1b or GT1b alters T cells and monocytes. GD1b caused decreased IL-6 and IL-10 production from CD4 T cells [110]. GT1b has a similar effect on monocytes [111]. When these ganglioside-treated T cells or monocytes are then co-cultured with B cells, antibody production is decreased. In contrast, GQ1b [112] and GD1a [113] can counteract the effects of GD1b and GT1b, respectively, and thus enhance Ig production by human PBMC. Meanwhile, GM2 and also GM3 gangliosides inhibit B cell production of IgG subclasses and IgM, with no effect on IgA subclasses [114]. Addition of TNF-α can counteract these inhibitions. Taken together, these studies show that GSL can negatively impact antitumor Ig production (see Figure 4, part “3”), but their effect on B cell antigen presentation has not been determined.
CD4 T cells, CD8 cytotoxic T cells, NKT cells. T cells play an important role in anti-tumor immunity, as helper cells for antibody production and cell-mediated immune responses as well as effector cells for tumor killing [115].
CD4 helper T cells. Type-1 T cell responses (represented by IFN-γ- and IL-2-producing T cells) are essential for the induction of cytolytic, anti-tumor immunity [116] while type-2 T cells producing IL-4, IL-6 and IL-10 are related to suppression of cytolytic activity [117]. In fact, cancer patients show increased type-2 cytokine production [118]. GSL influence T cell activation at numerous levels, including transcription of T helper cytokine genes, ligand-receptor binding of cytokines, antigen presentation, and T cell apoptosis (see Figure 4, part “4”). Rel/NF-?B regulate transcription of a large number of genes involved in T-cell development, maturation and proliferation [119]. Gangliosides derived from renal cell carcinomas (RCC) inhibit NF-?B activity in T cells, reduce IL-2 and IFN-γ expression [120] and increase apoptosis [121]. These changes are mediated by a decrease in the levels of RelA(p65), p50 and the antiapoptotic protein Bcl-xL (in a caspase dependent pathway), and can be impeded by blocking ganglioside synthesis [121].
When present during anti-CD3 antibody-induced activation of murine T cells, GT1b ganglioside reduces the levels of IFN-γ and increases IL-4, compatible with a shift towards a type-2 response [122]. Whereas the IFN-? reduction occurs in both CD4+ and CD8+ cell populations, the IL-4 enhancement is observed only in CD4+ T cells. However, the effects on the production of these two cytokines appear to be independent of each other. Similarly, human T cells activated in the presence of (RCC)- or brain-derived gangliosides show inhibition of IFN-γ (mRNA and protein) responses, apoptosis of activated T cells, but no influence on type-2 cytokines IL-4, IL-5 and IL-10 [123,124]. These findings are correlated in vivo, where T cells from RCC patients display a greater level of apoptosis. Interestingly, most of the apoptotic cells are GM2+ (GD2+ or GD3+) T cell populations, even though the mRNA expression levels for GM2 synthase are negligible [125]. Therefore, it is tempting to speculate that the ectopic presence of GM2 on apoptotic T cells is derived from the tumor, and that GM2 may be involved in triggering apoptosis. GSL-induced apoptosis is mediated by various mechanisms including reactive oxygen species, reduction of NF-?B-dependent antiapoptotic protein expression levels, induction of cytochrome c release and activation of caspases 3, 8 and 9, occurring on activated but not resting T cells [125-127]. Although opposed effects for some of these type-2-promoting changes are observed using more complex gangliosides such as GD1b, GT1b and GQ1b [128], this can be attributed to the usage of experimental conditions involving phytohemagglutinin (PHA)-stimulation of T cells. PHA engages multiple different glycosylated surface proteins, leading to activation of several, confounding different signaling pathways [129].
Antigen-specific activation of T cells requires the active participation of antigen presenting cells (APC) such as DC, macrophages and certain B cells, whose functions are affected by certain GSL. Ganglioside pretreatment of DC reduces antigen presentation to naïve T cells, leading to diminished proliferation and, accordingly, fewer IFN-γ-producing cells (type-1) and IL-4-producing cells (type-2) [106]. Among the molecular events implicated in antigen-specific activation of T cells, binding of IL-2 to its cell surface receptor (IL-2R) on T cells constitutes a critical step, in order to proceed through the G1 checkpoint of the cell cycle and trigger DNA synthesis and replication [130]. Brain-derived gangliosides can also interfere with these events by blocking IL-2 gene transcription in activated human T cells [124], and preventing phosphorylation of retinoblastoma protein (RB, a regulator of cell cycle progression) [124]. Moreover, mixtures of mono-, di- and tri-sialo gangliosides inhibit IL-2-stimulated proliferation of IL-2-dependent cell lines, CTLL-2 and HT-2 [131]. This dose-dependent effect occurs through direct binding of a lectin-like site on IL-2 to gangliosides, thus creating a competitive inhibition for IL-2 binding to IL-2R. A similar mechanism of direct cytokine-ganglioside interaction is thought to explain ganglioside-mediated inhibition of IL-4-stimulated helper T cell proliferation [132].
CD8 cytotoxic T cells. Cytotoxic T cells (CTL) mediate antitumor immunity by directly killing tumor cells [133]. The expansion of CTL is affected by GSL exposure. When splenocytes from mice immunized with FBL-3 erythroleukemia cells are rechallenged in vitro with irradiated FBL-3 tumor in the presence of gangliosides shed by these erythroleukemia cells, the tumor-specific response and the generation of CTL are suppressed. These GSL can impair responses to a secondary challenge as well (measured as reduced lymph node mass, cell number and proliferation), when coinjected in vivo with a primary FBL-3 cell immunization in mice [134]. An inhibition of the increase of draining lymph node T cells (CD3+) is also observed [134]. In addition, gangliosides can also influence the function of CTL (Figure 4, part “5”). Tumor-derived and individual highly purified gangliosides inhibit granule exocytosis-mediated cytotoxicity of alloantigen-specific and polyclonal CD8+ CTL [135]. Interestingly, they have no influence on lytic molecule expression, nor do they interfere with target cell recognition. Instead, they inhibit lytic granule release from CD8+ CTL by preventing TCR-induced lytic granule polarization, immunological synapse accumulation, and exocytosis [135].
Natural Killer T cells. Another cell type for which antitumor activity has been demonstrated is natural killer T (NKT) cells (Figure 4, part “6”). These T cells recognize a variety of both exogenous and endogenous lipid and glycolipid antigens presented in CD1 molecules, cell surface glycoproteins structurally resembling to MHC I molecules [136]. Upon activation, NKT cells produce cytokines that can influence immune responses against tumors [137]. Although GD3 can induce a GD3-reactive NKT cell response in melanoma [138], it is found to prevent NKT cell activation in ovarian cancer [139]. GM3 can also act as an inhibitory ligand for the NKT cell response to GD3 [140]. Asialo-GM2, shed from the murine T cell lymphoma line L5178Y-R, is another glycosphingolipid that prevents NKT cell recognition [141].
GSL are components of the external leaflet of plasma membranes and, as such, participate in numerous events of cellular interaction. Distinct GSL structures are generated through a series of intricate biosynthetic processes in the Golgi that are fine-tuned by several mechanisms. Tumors present several alterations in their GSL content. Equally important, GSL are actively released from the membrane of tumor cells and can bind immune cells to influence their function through multiple mechanisms, having a strong impact on impairing antitumor immunity and allowing tumor immune escape.
As auto-antigens present on normal cells at low levels, GSL are poorly immunogenic. Over several years, different strategies aimed at promoting anti-ganglioside immune responses have been studied, with disparate success. Besides the direct vaccination with GSL conjugated to adjuvants such as keyhole limpet haemocyanin [142], other approaches like anti-idiotypic antibodies [143], chimeric T cells [144], genetically-engineered antibodies [145] and GSL-mimicking peptides [146] have been assayed. These various strategies have been extensively reviewed elsewere [147,148].
Successful immune cell- or antibody-responses again GSL have the ability not only to target and kill tumor cells, but also to reduce the bioavailability of these molecules. Related to this concept, promising use of GSL synthesis inhibition in animal models of cancer [149-152] supports the notion that targeting GSL biosynthesis, for example with N-butyldeoxynojirimycin (a clinically approved inhibitor for glucosylceramide synthase), could be further explored in human cancer as a therapeutic approach, aiming to intervene on GSL metabolism of tumor cells and modulate GSL shedding, thus lessening the immunomodulatory effects of GSL.
This work was supported by National Institutes of Health (AR59126) and the Joseph B. Gould Foundation.