Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Research Article - (2012) Volume 2, Issue 2
Background: Influenza is a leading cause of morbidity and mortality in subjects with chronic diseases, who may also exhibit reduced immunogenicity to conventional influenza vaccines. MF59-adjuvanted influenza vaccine may enhance their immune response. Methods: We compared immunogenicity and safety of MF59-Adjuvanted Trivalent Influenza Vaccine (ATIV; Fluad®, Novartis Vaccines) and non-adjuvanted subunit (TIV; Agrippal®, Novartis Vaccines) in adults with at least one moderate to severe chronic condition. In this phase III, randomised, controlled, observer-blind study all subjects (18- 60 years of age) received one dose of ATIV (N=180) or TIV (N=179) vaccine during 2006/07 NH influenza season. Immunogenicity was tested using Hemagglutination Inhibition (HI) assay against vaccine and mismatched strains. Subjects were followed for safety for six months. Results: ATIV elicited significantly higher HI geometric mean titres (GMTs; P < .01) and mean-fold increases in titres (GMRs; P < .01) against all vaccine strains, compared with TIV. Seroprotection rates (HI ≥ 40) were 67–93% and 49–78% for ATIV and TIV groups, respectively (P < .01). ATIV also induced significantly higher GMTs against three mismatched strains (P < .05), and significantly greater GMRs against mismatched A strains (P < .05). Both influenza vaccines were well tolerated and safe, although ATIV elicited more solicited local and systemic (both 49%) reactions than TIV (both 28%). Most reactions (> 97%) were mild to moderate and all resolved spontaneously. Conclusion: ATIV is well tolerated, safe and confers higher and broader immunogenicity, when compared with a TIV, in adults with underlying chronic diseases.
Keywords: Adjuvant; Chronic diseases; Cross protection; Fluad; Influenza vaccine; MF59
AE: Adverse Event; CBER: Centre for Biologics Evaluation and Research; CHMP: Committee for Medicinal Products for Human Use; CI: Confidence Interval; CVD: Cardiovascular Disease; EMEA: European Agency for the Evaluation of Medicinal Products; GMR: Geometric Mean Ratio; GMT: Geometric Mean Titre; HI: Hemagglutination Inhibition; PP: Per Protocol; SAE: Serious Adverse Event
Influenza virus infects 5–15% of the population during a typical influenza season. The annual influenza epidemic worldwide results in about 3–5 million cases of severe illness and about 250 000–500 000 deaths (WHO Influenza Fact Sheet 221) [1]. Mortality is high in vulnerable populations, which include the elderly, very young children and those with underlying disease conditions compared with normal population [2,3].
Influenza infection causes a cascade of inflammatory response and can exacerbate underlying disease conditions, including Cardiovascular Disease (CVD) and diabetes, and can also lead to viral pneumonia, or a co-infection with other viruses or bacteria [4]. Epidemiological data indicates that risk for complications, hospitalization and death from influenza is higher for adults with chronic conditions including CVD. The WHO and national health authorities such as the US Advisory Committee on Immunization Practices (ACIP), therefore consistently recommend annual influenza vaccination for this group. Immunization is considered the best way of preventing influenza infection and its related complications. The increased risk of influenza related complications in adults with compromised immune systems makes it imperative to provide better prophylactic options and improve efforts for prevention.
The efficacy of influenza vaccine, however, depends largely on the degree of similarity between viral strains in the vaccine and those in circulation, as well as the age and the immuno-competence of the vaccine recipient. Vaccine formulations are updated each influenza season to match the strains that are predicted to be in circulation. However, mismatches can and do occur and this leads to diminished vaccine effectiveness. The addition of the adjuvant, MF59 to the vaccine formulation has been shown to enhance the cross-reactivity of induced antibodies against mismatched strains not included in the vaccine formulation, especially against the A/H3N2 strains that are epidemiologically prevalent in the adult community [5-7]. Moreover, studies have shown that MF59 can enhance the magnitude of immune responses in frail subjects, such as very young children, adults with underlying conditions, and the elderly, with an acceptable safety profile [8-10]. MF59 is an oil in water emulsion of the naturally occurring oil, squalene, and has been in use since 1997. Chronically ill subjects who might not be able to mount an adequate immune response could benefit from an MF59-adjuvanted vaccine.
Our study aimed to evaluate the immunogenicity, clinical tolerability and safety of MF59-adjuvanted and non-adjuvanted trivalent inactivated subunit influenza vaccines in adult subjects with underlying chronic diseases.
Study design
This phase III, randomised, observer-blind study was conducted at a single centre in Pianiga, Venice (Italy) from Nov 2006 to May 2007. The study protocols conformed to the ethical guidelines of the Declaration of Helsinki and Good Clinical Practice and were approved by the Ethics Committee of Merano, Venice.
A total of 330 subjects were planned to be randomised in a 1:1 ratio according to a computer generated randomisation list supplied by the study sponsor to receive a single intramuscular (IM) 0.5mL dose either of a subunit vaccine adjuvanted with MF59 (Fluad®, Novartis Vaccines [ATIV]) or of a conventional subunit vaccine (Agrippal®, Novartis Vaccines [TIV]). Both vaccines contained 15μg of hemagglutinin(HA) of each of the three influenza antigens recommended by the WHO for the Northern Hemisphere (NH) during 2006/07 influenza season: [A/New Caledonia/20/99 (H1N1)-like, A/Wisconsin/67/2005 (H3N2)-like and B/Malaysia/2506/2004-like].
Individuals eligible for enrolment in this study were male and female adult volunteers who were: (a) 18 to 60 years of age, mentally competent and able to comply with all the study requirements; (b) suffering from at least one of the following chronic diseases, i.e., moderate-to-severe hypertension, moderate-to-severe congestive heart failure, chronic obstructive pulmonary disease (COPD) or moderate-to-severe asthma, moderate-to-severe hepatic or renal insufficiency, arteriosclerotic disease or insulin-dependent diabetes mellitus.
Exclusion criteria included subjects who: (a) had experienced any acute disease within the 7 days prior to enrolment requiring systemic antibiotic or antiviral therapy or had experienced fever within the 3 days prior to enrolment (i.e., body temperature ≥ 38°C); (b) had a known allergy to any vaccine component; (c) had a history of neurological symptoms; (d) were pregnant, (e) had received more than one injection of influenza vaccine within 12 months prior to enrolment or had laboratory confirmed influenza within 6 months prior to enrolment; (f) had experienced an acute exacerbation of a COPD within 14 days prior to enrolment and any parenteral or oral cortical steroid or cancer chemotherapy/radiotherapy 60 days prior to enrolment and for the full length of the study. Counselling was provided and informed consent was obtained from each participant.
Assessment of clinical tolerability and safety
Subjects were observed for 30 minutes after injection for any immediate reactions. Local reactions such as ecchymosis, erythema, induration, swelling and pain at injection site, as well as systemic reactions including arthralgia, chills, fever, headache, malaise, myalgia, fatigue and sweating were recorded on a diary card by the subjects during the 7 days post-vaccination. Any serious or non-serious adverse event (AE) occurring during the 3 weeks post-vaccination were recorded. All serious adverse events (SAEs) were collected in the six months follow-up (up to day 181 after vaccine injection).
Assessment of immune responses
Blood samples were obtained from each subject before vaccination and at 3 weeks post-vaccination. Haemagglutination Inhibition (HI) antibody titres were measured by Clinical Southern Research Institute (Birmingham, AL, USA). Immunogenicity against the three vaccine strains, [A/New Caledonia/20/99 (H1N1)-like, A/Wisconsin/67/2005 (H3N2)-like and B/Malaysia/2506/2004-like], was determined using the following parameters: geometric mean titres (GMT) with 95% confidence intervals (CI); geometric mean ratios (GMRs) of post to prevaccination titres; seroprotection rate, defined as the percentage of subjects achieving an HI titre≥40; and seroconversion rate, defined as the percentage of subjects achieving either at least a fourfold increase in HI titre from a non-negative pre-vaccination titre [≥10] or a rise from <10 to ≥40 in those who were seronegative at baseline. The results were assessed according to the Committee for Medicinal Products for Human Use (CHMP; formerly CPMP) criteria for approval of influenza vaccines in healthy adults.
Sera collected during the trial were also tested against mismatched A/H3N2 (A/New York/55/2004-like) and B (B/Jiangsu/10/2003-like) strains recommended for 2005/06 NH vaccination campaign, and against the A/H1N1 strain (A/Solomon Islands/3/2006-like) that emerged during subsequent influenza season (2007/08).
Statistical methods
Data were analysed using SAS version 9.1.3 (SAS Institute, Cary, NC). Immunogenicity was analysed for the per protocol population defined as all subjects who received the vaccine dose correctly, provided serum samples at both scheduled time points and had no major protocol violations. Statistical significance between pre- and post-vaccination titres was assessed using ANOVA with one factor for vaccine group. Titres observed in different vaccine groups were also compared using ANOVA with one factor for vaccine group. The chi-square test was performed to analyse differences between proportions of subjects. A P value<.05 was considered as statistically significant.
Study population
A total of 361 adult subjects were enrolled and randomised, while 359 received either ATIV (N=180) or TIV (N=179) influenza vaccine and were included in the clinical tolerability and safety analysis. Of these, 163 in the ATIV group and 156 in the TIV group completed the study according to the protocol, and provided a sufficient blood to be included in the immunogenicity analysis. The demographic and other baseline characteristics were balanced between the two vaccine groups (Table 1). More than half of enrolled subjects had not previously received influenza vaccine (61% ATIV and 63% TIV).
| Variable | Sub/MF59 | Subunit |
|---|---|---|
| N=181 | N=180 | |
| Mean Age (yrs ± SD) | 49.2 ± 10.0 | 49.7 ± 9.2 |
| Male/Female (%) | 106/75(59%/41%) | 89/91(49%/51%) |
| Mean Weight (kg ± SD): | 78.84 ± 15.75 | 77.07 ± 16.03 |
| Mean Height (cm ± SD): | 170.1 ± 8.4 | 167.3 ± 9.1 |
| Body Mass Index:( ± SD): | 27.20 ± 4.84 | 27.45 ± 4.92 |
| Previously Vaccinated: | 71 (39%) | 67 (37%) |
| Chronic Medical Condition – n (%) | ||
| Moderate to Severe Hypertensiona | 132 (73%) | 134 (74%) |
| Only Hypertension | 112 (63%) | 103 (60%) |
| Moderate to Severe Congestive Heart Failurea | 10 (6%) | 13 (7%) |
| COPD or Moderate to Severe Asthmaa | 45 (25%) | 40 (22%) |
| Moderate to Severe Hepatic or Renal Insufficiencya | 11 (6%) | 15 (8%) |
| Arteriosclerosis or Diabetes Mellitusa | 8 (4%) | 10 (6%) |
| 2 co-morbidities | 12 (7%) | 25 (15%) |
| 3 co-morbidities | 6 (3%) | 2 (1%) |
Table 1: Demography and Baseline Characteristics – Enrolled Population.
Immunogenicity
Serologic analysis vs. homologous (vaccine) and heterologous (mismatched) strains was performed on 319 subjects, 163 in the MF59-adjuvanted vaccine group and 156 in the conventional TIV vaccine group.
Immune responses Vs vaccine strains
A/H3N2 Antigen (A/Wisconsin/67/2005-like): The pre-vaccination GMTs were similarly low in both vaccine groups. Significantly higher GMTs (351 vs. 159, P<.001) and GMRs (23 vs. 13, P=.007, Figure 1a) were recorded in the ATIV group compared with the TIV group. Furthermore, 77% of subjects in the ATIV group achieved seroconversion or significant increases in HI antibody titres, compared with 60% in the TIV group (P=.001). Post-vaccination, the seroprotection rate was significantly greater in the MF59-adjuvanted vaccine group (93 % vs. 78%, P<.001; Figure 2).
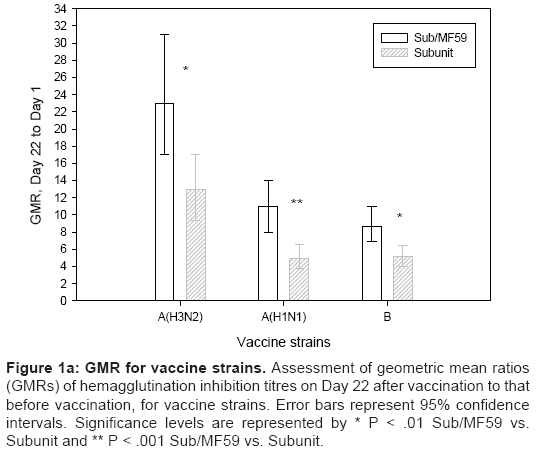
Figure 1a: GMR for vaccine strains. Assessment of geometric mean ratios (GMRs) of hemagglutination inhibition titres on Day 22 after vaccination to that before vaccination, for vaccine strains. Error bars represent 95% confidence intervals. Significance levels are represented by * P < .01 Sub/MF59 vs. Subunit and ** P < .001 Sub/MF59 vs. Subunit.

Figure 1b: GMR for mismatched strains. Assessment of geometric mean ratios (GMRs) of hemagglutination inhibition titres on Day 22 after vaccination to that before vaccination, for mismatched strains. Error bars represent 95% confidence intervals. Significance levels are represented by * P < .05 Sub/MF59 vs. Subunit and ** P < .01 Sub/MF59 vs. Subunit.
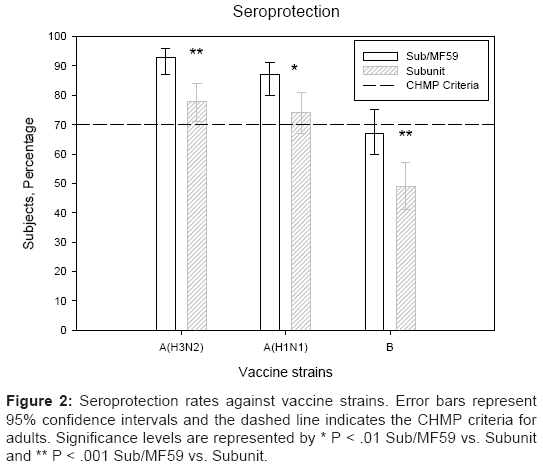
Figure 2: Seroprotection rates against vaccine strains. Error bars represent 95% confidence intervals and the dashed line indicates the CHMP criteria for adults. Significance levels are represented by * P < .01 Sub/MF59 vs. Subunit and ** P < .001 Sub/MF59 vs. Subunit.
A/H1N1 Antigen (A/New Caledonia/20/99-like): The pre-vaccination GMTs were similar in both groups. Significantly higher GMTs (174 vs. 89, P<.001 and GMRs (11 vs. 4.97, respectively; P<.001; Figure 1a) were recorded in the MF59-adjuvanted group compared with the non-adjuvanted vaccine group. At three weeks after vaccination 63% vs. 47% of subjects achieved seroconversion against the A/H1N1 strain (P=.005) and seroprotection rates were 87% vs. 74% in the two vaccine groups, respectively (P=.006; Figure 2).
B Antigen (B/Malaysia/2506/2004-like):The pre-vaccination GMTs were similarly low in both vaccine subgroups. Significantly higher HI antibody titres (57 vs. 33, P=.001) and GMR (8.72 vs. 5.12, P=.001; Figure1a) were recorded in ATIV vs. TIV. Three weeks after vaccination, 67% and 49% of subjects achieved seroprotection (P<.001; Figure 2), while 62% and 44% of subjects achieved seroconversion rates, respectively (P=.002). The immunogenicity results are summarised in Table 2.
| Sub/MF59 (N=163) | Subunit (N=156) | |||||
|---|---|---|---|---|---|---|
| STRAINS | A(H3N2) | A(H1N1) | B | A(H3N2) | A(H1N1) | B |
| PRE-VACCINATION | ||||||
| GMT | 15 | 17 | 6.58 | 12 | 18 | 6.41 |
| 95% CI | 12-19 | 13-21 | 5.97-7.25 | 9.88-16 | 14-23 | 5.8-7.09 |
| Seroprotection ratea % | 28 | 31 | 6 | 23 | 37 | 4 |
| 95% CI | 21-36 | 24-38 | 3-11 | 17-30 | 29-45 | 2-9 |
| POST VACCINATION | ||||||
| GMT | 351** | 174** | 57* | 159 | 89 | 33 |
| 95% CI | 269-457 | 135-224 | 45-72 | 121-208 | 68-115 | 26-42 |
| Seroprotection ratea % | 93** | 87* | 67** | 78 | 74 | 49 |
| 95% CI | 87-96 | 80-91 | 60-75 | 71-84 | 67-81 | 41-57 |
| Seroconversionb % | 77* | 63* | 62* | 60 | 47 | 44 |
| 95% CI | 70-83 | 55-70 | 54-69 | 52-68 | 39-55 | 36-52 |
Table 2: Immunogenicity Results (Vaccine strains) – Per Protocol (PP) population.
Immune responses vs. mismatched strains
As a further analysis the immune responses induced by ATIV and TIV vaccine against mismatched A/H3N2 and B strains recommended for 2005/06 NH vaccination campaign, and against the A/H1N1 strain that emerged during 2007/08 influenza season were studied.
A/H3N2 Antigen (A/California/7/2004-like): Baseline GMTs were similar in both vaccine groups (32 vs. 26), and post-vaccination significantly higher GMT (676 vs. 355, P<.001) and GMR (21 vs. 14, P=.048; Figure 1b) were observed in the ATIV group compared with the TIV group. A total of 74% of subjects achieved seroconversion or a significant increase in HI antibody titres in the ATIV group compared with 66% in the TIV group. Seroprotection rates were 96% vs. 91%.
A/H1N1 Antigen (A/Solomon Islands/3/2006-like): Pre-vaccination GMTs were similar and low in both groups, but post-vaccination significantly higher GMTs (73 vs. 45, P=.013) and GMR (6.58 vs. 3.89, P=.004; Figure 1b) were recorded for ATIV compared with the TIV group. Seroconversion was 48% vs. 35% (P=.018) and the seroprotection rates were 67% vs. 56%, respectively.
B Antigen (B/Shanghai/361/2002-like): Baseline GMTs were similarly low in both the vaccine groups. After vaccination GMTs were significantly higher (45 vs. 31, P=.022) for the ATIV group and GMRs were recorded as 4.05 vs. 3.16 (Figure 1b). A total of 40% subjects achieved seroconversion in ATIV vs. 35 % in the TIV group. Seroprotection rates were 62 % and 53 % respectively.
Clinical tolerability and safety
All 359 subjects who were vaccinated (180 in ATIV, and 179 in TIV group) were included in the safety analysis set. The percentages of subjects reporting solicited local reactions in the ATIV group were higher than in the TIV group (49% and 28%, respectively). The most frequent local reaction was pain, experienced by 81 (45%) ATIV and 36 (20%) TIV recipients (P<.001; Table 3). In both vaccine groups solicited local reactions were generally of mild intensity and short lived, generally resolving within 2 or 3 days following vaccination and none continued beyond Day 7.
| Type of Reaction | Number (%) of Subjects with Solicited Reactions | ||||
|---|---|---|---|---|---|
| Sub/MF59 N=180 | Subunit N=179 | ||||
| Any reaction | 112 (62%) | 78 (44%) | |||
| Local reaction | (a Severe: > 100 mm) | 89 (49%) | 50 (28%) | ||
| Ecchymosis | Any | 13 (7%) | 14 (8%) | ||
| Severea | 0 | 0 | |||
| Erythema | Any | 7 (4%) | 14 (8) | ||
| Severea | 0 | 0 | |||
| Induration | Any | 24 (13%) | 15 (8%%) | ||
| Severea | 0 | 0 | |||
| Swelling | Any | 15 (8%) | 8 (4%) | ||
| Severea | 0 | 0 | |||
| Pain* | Any | 81 (45%) | 36 (20%) | ||
| Severe | 4 (2%) | 1 (1%) | |||
| Systemic reaction | 89 (49%) | 50 (28%) | |||
| Chills | Any | 15 (8%) | 17 (9) | ||
| Severe | 3 (2%) | 1 (1%) | |||
| Malaise** | Any | 40 (22%) | 23 (13%) | ||
| Severe | 3 (2%) | 2 (1%) | |||
| Myalgia* | Any | 55 (31%) | 17 (9%) | ||
| Severe | 5 (3%) | 2 (1%) | |||
| Arthralgia | Any | 33 (18%) | 23 (13%) | ||
| Severe | 4 (2%) | 2 (1%) | |||
| Headache | Any | 37 (21%) | 30 (17%) | ||
| Severe | 4 (2%) | 4 (2%) | |||
| Sweating | Any | 10 (6%) | 8 (4%) | ||
| Severe | 2 (1%) | 1 (1%) | |||
| Fatigue | Any | 27 (15%) | 24 (13%) | ||
| Severe | 4 (2%) | 2 (1%) | |||
| Fever (axil.temp.) | ≥38.0°C | 1 (1%) | 0 | ||
| ≥ 40°C | 0 | 0 | |||
| Other indicators of reactogenicity | |||||
| Stayed home due to reaction | 7/179 (4%) | 2/178 (1%) | |||
| Analgesics/antipyretics use | 18 (10%) | 11 (6%) | |||
Table 3: Overview of Subjects with Solicited Reactions.
The most commonly reported solicited systemic reactions were myalgia (ATIV 31%, TIV 9%; P<.001), malaise (ATIV 22%, TIV 13%; P=.02) and headache (ATIV 21%, TIV 17%). In both vaccine groups solicited systemic reactions were generally mild or moderate in severity, with severe reactions reported by 1–3% across groups and reactions. They were mostly experienced within 3 days following vaccination and resolved by Day 7.
Unsolicited adverse events (AEs) were experienced by 18% of ATIV and 20% of TIV recipients, but only 4 (2%) subjects in each group experienced AEs considered possibly/probably related to study vaccine: vertigo (1 ATIV, 2 TIV), 2 parathesia (both ATIV), 1 burning sensation (ATIV), 1 headache and 1 hot flush (both TIV). No death occurred during the study. SAEs were reported by four subjects (two ATIV; moderate dyspnea, sarcoma relapse, and two TIV; mild syncope, colon cancer). None was assessed by the investigator as related to the study vaccines.
Annual circulation of the influenza virus coincides with a significant seasonal increase in morbidity and mortality, resulting from both the symptoms of influenza itself and from other associated illnesses. For example, one study has estimated a rate of 115 hospitalisations per 100,000 person-years for circulatory and respiratory illness associated with influenza[3]. Other studies have shown that mortality from ischaemic heart disease, acute myocardial infarction, cerebrovascular disease, diabetes, cardiorespiratory disease and chronic obstructive pulmonary disease (COPD) was associated with influenza [11-14].
Adults with underlying chronic conditions are at increased risk of developing influenza-related complications, but a considerable percentage remain unvaccinated. Influenza infection has been suggested to be associated with a transient increase in the risk of vascular events [12][15]. The risk for cardiac events is demonstrated by the fact that vaccination helps to prevent the cascade of inflammation, plaque instability and rupture induced by infection [16]. In other studies influenza vaccination has been linked with reduced risk for stroke, diabetes and other chronic disease conditions [17] [4] [18]. Moreover, the predominance in recent influenza seasons of the A/H3N2 strain, which generally causes a higher number of serious illnesses and hospitalisations than influenza A (H1N1) or influenza B, underscores the need for better protection. Effectiveness of conventional influenza vaccines is substantially lower in the vulnerable as compared with healthy adults [9]. The development of adjuvanted influenza vaccines, to improve the effectiveness of influenza vaccines in the frail subjects, may improve protection in these groups. Vaccination with MF59-adjuvanted vaccine resulted in an enhanced immune response in frail populations, such as the elderly and subjects with underlying chronic disease, compared with a conventional TIV [9,19]. Furthermore, a cross-reactive immune response has been observed, demonstrating the vaccine’s ability to confer protection against a broader range of influenza virus strains [5,20,21]. More than 50 million doses of the MF59-adjuvanted seasonal influenza vaccine Fluad have been distributed worldwide since first licensure. Overall the adjuvanted vaccine is well tolerated and safe, although there is a clear increase in injection site reactions (especially injection site pain), but these are usually of mild intensity and short living (within 2-3 days from injection) [22].
Our study aimed to investigate the immunogenicity and safety of MF59-adjuvanted and a conventional non-adjuvanted TIV vaccine in adults (18–60 years) with chronic medical conditions, a target group for influenza vaccination. The immune response elicited by MF59-adjuvanted vaccine showed significantly higher GMTs, GMRs, seroprotection and seroconversion rates for vaccine strains tested compared with the TIV vaccine group. Additionally, analysis against heterologous (mismatched) strains showed that MF59-adjuvanted vaccine consistently induced significantly higher HI antibody titres, more subjects achieving seroprotection and seroconversion against heterologous strains compared with TIV vaccine. These findings are consistent with those previously reported in literature, which have shown that MF59 adjuvant induces a significantly higher cross reactive immunogenicity against drifted strains [6,21,23]. As noted, both vaccines were well tolerated and safe in adults with chronic diseases with the known increase in incidence of mild, short-lived local reactions was observed for ATIV, consistent with previous studies. A large integrated safety analysis supports the good safety profile of the adjuvant and of MF59-adjuvanted influenza vaccines [24] and the potential benefits of enhanced protection outweigh the transient injection site discomfort.
To conclude, in adults with underlying chronic diseases the MF59-adjuvanted subunit influenza vaccine showed a superior immunogenicity versus both vaccine and mismatched strains, and especially the A/H3N2 that is epidemiologically relevant in adult subjects, when compared with a non-adjuvanted TIV. Administering a more immunogenic influenza vaccine is generally desirable and will be especially beneficial for people with underlying chronic conditions, who are at higher risk of influenza-related complications.
M.P., P.N., DT O’Hagan, N.G. are employees of Novartis Vaccine and Diagnostics. V.B. was the co-ordinating investigator, G.A. was the principal investigator and T.B., C.B., C.G., I.M., P.M. were study investigators. No other disclosures were reported.
Vincenzo Baldo participated to conceive and design the study, to collect data, and in the overall coordination and drafting of the manuscript. Michele Pellegrini contributed to the study design, to the interpretation of results and the draft and review of the manuscript. Tatjana Baldovin participated in the study conception and design and in data collection, participated in the discussion and review of the manuscript. Gabriele Angiolelli participated in the study conception and design and in data collection, participated in the discussion and review of the manuscript. Pantaleo Nacci provided statistical and programming support for the data analysis and participated in the discussion and review of the manuscript. Derek O’Hagan has generated the data for the heterologous activity and participated in the discussion and in the interpretation of results. Nicola Groth contributed to the study design, to the interpretation of results and the review of the manuscript.The Family Medicine Group of Pianiga (formed by the MD) has enrolled the subjects for the clinical trial.
The authors wish to thank Anke Hilbert, Ralf Jaeger and Francesca Ballini (Novartis Vaccines and Diagnostics) for their support during the serological and statistical analyses. The authors would also like to acknowledge Shruti Priya Bapna (Novartis Vaccines and Diagnostics) for support with the manuscript preparation.
ClinicalTrials.gov NCT00519064.