Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2013) Volume 6, Issue 8
Background: High throughput analysis like mass spectrometry dedicated to clinical proteomics offers new insights into clinical research. This promising technology generates high-dimensional datasets with a huge amount of biological input. Working with these high-dimensional datasets has created challenges for statistical methods and there are still weaknesses in current statistical analysis that have to be overcome to get an accurate interpretation of “omics” studies. The central question is that of a reliable identification of new prognostic and diagnostic biomarkers. Although observed in previous studies, these mechanisms of identification and validation of new markers have been inadequately explained and often dissociated.
Results: The aim of our study was therefore to show how candidate markers are sometimes selected in identification studies because of biased estimations of their effect. To achieve this goal, this work was conducted through the simulation of high-dimensional studies concerning survival. We showed how the selection mechanism involved in identification studies influences a mechanism called regression to the mean. This in turn leads to a biased estimation of the effect size and thus to optimism when considering validation studies.
Conclusions: This study demonstrated why the discovery of new robust markers is only possible through well-designed studies relying on consistent sample sizes for the identification step. Due to the above mentioned mechanisms of identification and validation, pertinent candidate biomarkers in high-dimensional clinical studies require non-biased estimation, and this right from the identification step. Only then will it lead to consistent studies and thus reach benefit in terms of health care.
Keywords: High dimension; Biomarker selection; Validation; Regression to the mean
Nowadays, cancer research is making use of new high throughput technologies, like mass spectrometry in the field of clinical proteomics. Mass spectrometry signals show the proteomic profiles of the individuals under study at a given time. These profiles correspond to the recording of a large number of proteins, much larger than the number of individuals. Thus, this leads to the generation of highdimensional datasets with a huge amount of biological input. Working with high-dimensional datasets has created a number of challenges for statistical methods. In particular, it raises two main statistical questions: the identification of candidate markers and their validation in further studies.
A classical clinical study starts with an a-priori hypothesis made by the clinician about the potential prognostic or diagnostic effect of one particular clinical factor. Usually, only one or very few variables are tested in a single study. Statistical models aim to validate (or not) this hypothesis by estimating the strength of the association between this variable and the clinical outcome of interest, and by testing its significance. In contrast, there is no a-priori hypothesis in “omic” studies, where a huge number of variables are tested simultaneously. Thus, a two-step strategy is needed for these studies.
The first step corresponds to identification studies designed to select a list of candidate biomarkers tested among a high number of biological parameters; this is conducted by analyzing a sample of the population under study. This identification step can in turn be broken down into two sub-steps: estimation and selection. In fact, the selection of relevant markers relies on the estimation of the strength of association between each biological input and the clinical outcome of interest. Only values with a significantly high enough strength of association are selected.
These studies lead to the acquisition of large number of variables. These variables are potential biomarkers and may be of several types. In the context of clinical proteomics, data generated by mass spectrometers correspond to the proteomic profile of each of the individuals under study at a given time. These profiles correspond to the recording of the intensities of a high number of proteins expressed by the genome of the individuals. Besides proteomics datasets, other “omics” datasets representing other biological levels are also concerned by this huge quantity of variables. Genomics aims at learning about genes through the study of SNPs for example (Single Nucleotide Polymorphism, i.e. DNA sequence variation), while transcriptomics aims at learning the expression and regulation of genes through the study of RNA. All these studies are characterized by the same huge amount of variables. Given the cost or the difficulty to get biological samples, this large number of variables often go with relatively low number of observations. By chance alone, many potential markers may be found significantly associated with the outcome, even though most of them may not actually be linked to diagnosis or prognosis. The question of multiple testing was hot debated through the definition of the False Discovery Rate (FDR), for example, that is, the expected proportion of false positives among the genes declared as significant [1]. When looking for differential genes, Pawitan et al. [2] showed that the FDR is mostly influenced by the proportion of truly differentially expressed genes, and by the sample size. Some additional works showed how the type I error rate was related to the power in high-dimensional setting [3,4], the increase in the power being at the cost of the FDR. Some authors illustrated the issues related to the selection process in the “omic” field. For example, Michiels et al. [5] showed through the well-known dataset from Van’t Veer et al. [6], that the list of selected candidates was highly unstable and depended on the composition of the identification set. Later, Ein-Dor et al. [7] proposed a tool to evaluate the selection process and showed that thousands of samples are needed to ensure a robust signature.
The second step corresponds to validation studies designed to confirm the previously selected candidate(s) as biomarker(s). This step can also be broken down into two sub-steps: re-estimation and confirmation. These studies aim to re-estimate on independent datasets the strength of association of the previously selected candidates, and thus confirm (or invalidate) the relevance of candidates as markers. Relevant markers may have optimal predictive quality. Some authors illustrated the divergence between the strength of association estimated in identification studies and that estimated in validation studies. In particular, Michiels et al. [5] showed on several well-known datasets how an inadequate validation led to the publication of overoptimistic results compared with those from their own analyses. Later, Truntzer et al. [8] showed how high-dimensional data analysis was subject to greater optimism-that is to say an over-estimation of the strength of association- compared with analysis of classical clinical variables.
Some solutions were proposed to correct the optimism bias linked to the selection process. Using resampling methods [9,10] or penalized regression [11-13] are such examples. The objective of this paper, however, is not to propose new solutions, but rather to explain how it works. Indeed, to our knowledge, the questions of identification and validation have been highlighted, but neither the mechanisms, nor the ways in which these two steps are strongly associated have been thoroughly explained. In this work, we propose to explain the link between estimation and selection. To understand the process involved, we will analyse how candidate markers are selected in identification studies and how their estimated strength of association may be reduced-and thus not confirmed-when re-estimated in validation studies. In other words, to better understand how selection leads to optimism, we propose to show how the estimation bias that may occur in the identification steps leads to selection of inappropriate candidate markers. For recall, regression toward the mean refers to the phenomenon that a variable that is extreme on its first measurement will tend to be closer to the mean of the distribution on a later measurement [14,15]. In fact, let consider a given variable. Its measurement varies around its mean following a given distribution. When sampling a first measurement from this distribution, there is a low probability of observing it extreme. So, if a first measurement is extreme, there is high probability that the second one spontaneously regress towards the mean value [16].
Simulation of the datasets
Comprehension of the mechanisms involved in the identification and validation steps was achieved through simulations of survival “omic” datasets. Indeed, simulations have main advantage of offering a situation in which the truth is known and can even be controlled. The same processes as those encountered in real-life clinical studies can also be reproduced with the advantage that all the parameters can be controlled.
The same simulation process as described in a previous paper by the same authors was used [8]. Here is a brief description of this process. A classical way to link variables to censored survival data is to use the Cox proportional hazards model. Let us de ne X an (n, p) matrix of p variables for n individuals. For each of the n individuals, the follow-up times were noted t1,...., tn as were the event-indicators δ1,....,δn with δi=1 if the event occurred and δi=0 if it did not occur. At time t, the Cox proportional model is given by
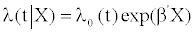 (1)
(1)
Where λ0(t) is a baseline hazard function, β={β1,...,βn} is the vector of parameters and X1,....,Xp are the vectors of length n describing each of the p variables for the n patients.
We simulated a virtual population of size n in which each individual is described by p “omic” variables -with n<

Figure 1: Flowchart of the whole process conducted in this study. Representation of the whole process involved in the discovery of new biomarkers as simulated in this study. The “truth” left part of the figure corresponds to the simulation settings representing the global population. A total of p=1000 variables were under study, among whom p0 were under the null hypothesis H0 of no association with survival, and p1=20 under the alternative hypothesis. The right part of the figure corresponds to the identification and validation steps occurring in “omic” studies. R variables are selected on the identification set, and then validated in 50 distinct validation sets.
For a fixed set of parameters p and p1, 200 identification sets of n patients were simulated according to the above design. For each of these identification sets, 50 corresponding validation sets were drawn up following the same design. This overall process was performed by considering n in {100,200,400,1000}. In this study we chose p=1000 and p1=20.
For each simulated identification set, univariate Cox regression models were used to estimate the strength of the association of each variable through survival model parameters. Based on these estimations, the R most contributive variables were selected in a univariate way.
Selecting variables in the multiple hypothesis setting results in considering the problem of testing simultaneously p null hypotheses, leading to different situations, described in Table 1 [17]. Among the p corresponding variables, p1 are under the H1 hypothesis 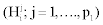 , while p0 are under the H0 hypothesis
, while p0 are under the H0 hypothesis 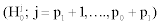 . The test leads to the rejection of R hypothesis.
. The test leads to the rejection of R hypothesis.
| # Not rejected | # Rejected | # Total | |
|---|---|---|---|
| # True hypothesis | U | V | p0 |
| # Not true hypothesis | T | S | P1 |
| # Total | p-R | R | p |
U: True negatives; T: False negatives; V: False positives; S: True positives.
Table 1: Multiple testing setting.
Among the R rejected hypotheses, V are under the null hypothesis (False Positives or FP), whereas S are actually under the alternative hypothesis (True Positives or TP). In the same way, among the (p-R) variables, T were wrongly not selected. The Type I error concept had to be newly defined to take into account the huge number of hypotheses tested. The basic idea is to adjust p-values of usual test statistics in order to control the global error rate. For this purpose, the control of the False Discovery Rate, that is the expected proportion of Type I errors among the rejected hypotheses FDR = E(V R), is commonly used [1]. In general, one would like to minimize the number V of false positives, or Type I errors and the number T of false negatives, or Type II errors, thus maximizing the power, defined as 1 E(S) p .
Note that the identity of the R variables depends on the identification set. Indeed, the same set of variables is not systematically selected from one dataset to the other.
• In this work we were interested in the estimation of the strength of association for the R variables, which was estimated through univariate Cox survival models. Once the R variables had been selected on one identification set, the Cox coefficients of these R variables were then re-estimated on the corresponding validation sets. Estimations for the V and the S variables were stored separately. Note that there was no new variable selection on the validation sets. In parallel, Cox coefficients for the p0 and p1 variables were estimated on each identification set. The whole process was performed for each of the 200 identification sets, and is illustrated in Figure 1. To sum up, we considered the distributions of the Cox coefficients estimated for: The p, p0 and p1 variables over the 200 identification sets. The sets of p, p0 and p1 variables are respectively denoted Ωp, Ωp0 and Ωp1, hereafter.
• The V, S and R variables over the 200 identification sets. These sets of variables are respectively denoted ΩV,ΩS and ΩR, hereafter. The ΩV, ΩS and ΩR sets were defined separately on each identification set. Keep in mind that the variables constituting these sets of variables are not the same, depending on the identification dataset. While R depends on the datasets when the FDR control is applied, it is fixed otherwise. The distributions of the estimates for these sets of variables will make it possible to understand what happens in identification studies where p “omic” variables are tested, without a-priori hypotheses about their relationship with survival.
• The V, S and R variables over 50 validation sets for each of the 200 identification sets. We insist on the fact that ΩV,ΩS and ΩR were not newly defined on the validation sets. At this step, the selection process is over, and the corresponding validation studies are conducted. We also remind the reader that the identification and the validation sets are the same size.
Results
The comparison of the above described densities were used to illustrate how the selection mechanism involved in identification studies influences regression to the mean, and how it leads to overestimation of the strength of association and thus to optimism.
Results are shown through histograms that display the density of each of the distributions of interest. Each of the following figures is related to one particular set of variables (Figure 1) from the last, but one column of Table 1. Whatever the figure, each of the four panels was obtained with a specific sample size, with n={100; 200; 400; 1000}. The vertical line with abscissa 0.2 corresponds to the simulated strength of association; in other words, it corresponds to the mean distribution of the Ωp1 estimates. Note that the number of estimates contributing to the distributions density is not the same for the identification and validation sets. In the following, results concern the “top-20” approach.
Figure 3 concerns distributions of the strength of association estimated over 200 identification sets for the Ωp0 (grey histograms) and ΩV (horizontal hatching) sets of variables. Let us restate that ΩV variables correspond to FP, and are also a subset of Ωp0. The distribution of the estimates for the Ωp0 variables is around 0 (its true mean) and gets narrower with increasing sample sizes. The distribution of the estimates for the ΩV variables is bimodal. Variables from the ΩV set were selected in both extremes of the Ωp0 estimates distribution. With n=100 or 200, the estimates of the variables selected in the right extreme are even higher than the true strength of association simulated for variables related to survival (vertical line). In parallel, one observes that each mode of the distribution of the estimates for ΩV is far away from the mean distribution of Ωp0 estimates. When increasing the sample size, the distribution of Ωp0 estimates gets narrower and thus the extreme of the distribution moves away from the vertical line. In parallel, each mode of the distribution of the estimates for ΩV approaches zero.
To go further, Figure 4 illustrates the mechanism encountered when considering the estimates of all selected variables constituting ΩR. This figure concerns both identification and validation steps. For this purpose, each panel shows the distribution of the estimated strength of association for ΩR computed over 200 identification sets parameters (horizontal hatching) and over 200*50 validation datasets (diagonal hatching). The vertical dotted line corresponds to the mean distribution of ΩR estimates computed on the validation sets. For the identification datasets (horizontal hatching), the distribution started from bimodal with n=100 to unimodal with n=1000 individuals. With small sample sizes, the modes of the distribution are far from 0.2. With n=1000, the left mode vanishes and the mean distribution of the estimates for ΩR tends toward the mean distribution of the estimates for Ωp1, that is 0.2. For the validation sets, the distribution is first unimodal (n=100), and tends toward 0, indicating that the majority of the ΩR variables are in reality false positives. With n=200, a shoulder appears on the left of the distribution, and the shoulder moves from the left to the right. In parallel, the mean distribution of the strength of association estimated for ΩR tends towards 0.2 and matches this value with n=1000. Another noticeable observation is that estimates of R from the identification and the validation sets come closer with increasing sample sizes. With n=1000, the estimations obtained on identification and validation sets join.
With too few individuals under study, estimates for Ωp1 (Figure 2) and Ωp0 (Figure 3) are highly fluctuating, which is indicated by the wide distributions. Variables are selected because of their high estimates, which lie in the extreme of the distributions of the estimates for the Ωp0 or Ωp1 variables. As a consequence, the mean estimates obtained for selected variables were far from their true means: the means for ΩV and ΩS were higher than the means for Ωp0 and Ωp1, respectively. This demonstrates a selection bias. When increasing the sample sizes, the mean distribution of the estimates for ΩS tends toward the mean distribution of the estimates for Ωp1 due to a decrease in the selection bias. This is due to the regression to the mean phenomenon, which is influenced by the selection process. This phenomenon affects both Ωp0 and Ωp1 estimates with different consequences. As for Ωp1, regression to the mean leads to over-estimation of the strength of association for true positives. As for Ωp0, the poor distribution of its estimates leads to the inappropriate selection of some FP variables that have in fact no effect on survival.

Figure 2: Parameter estimates with varying n for variables under the H1 hypothesis. This figure only concerns estimates for identification sets. Each of the four panels was obtained with a specific sample size with n={100; 200; 400; 1000}. Whatever the panel, the following distributions were plotted: 1-distribution of the estimates for the Ωp1 variables computed over 200 identification sets (grey histogram). 2-distribution of the estimates for the ΩS set of variables computed over 200 identification sets (histogram with horizontal hatching). The vertical continuous line indicates 0.2.
With n=100, estimates for Ωp1 are highly fluctuating, as shown by the wide distribution. Variables are selected in the extreme of the distributions of the estimates for Ωp1, and these mean estimates are thus far from their true means. When increasing the sample sizes, the mean distribution of ΩS estimates tends toward the mean distribution of Ωp1 estimates. This is an illustration of the regression to the mean phenomenon that leads to over-estimation of the strength of association for true positives.

Figure 3: Parameter estimates with varying n for variables under the H0 hypothesis. This figure only concerns estimates for identification sets. Each of the four panels was obtained with a specific sample size with n={100; 200; 400; 1000}. Whatever the panel, the following distributions were plotted: 1-distribution of the estimates for the Ωp0 variables obtained over 200 identification sets (grey histogram). 2-distribution of the estimates for the ΩV sets obtained over 200 identification sets parameters (histogram with horizontal hatching). The vertical continuous line indicates 0.2.
With n=100, estimates for Ωp0 are highly fluctuating, as shown by the wide distribution. Variables are selected in the extreme of the distributions of Ωp0 estimates and the mean estimates of ΩV variables are thus far from their true means. When increasing the sample sizes, the distribution of the estimates for the Ωp0 variables gets narrower and the mean distribution of the ΩV variables estimates decreases. This illustrates the regression to the mean phenomenon that leads to the inappropriate selection of some FP variables that have in fact no effect on survival.
In the light of these comments, Figure 4 shows how the above cited mechanisms affect the selection of candidate biomarkers, and the further consequences in terms of their confirmation. Because of the regression to the mean phenomenon described above, R is a mixture of S and V. Whatever the dataset (identification or validation), the right mode mostly consists of the estimates of true positives (ΩS), whereas the left mode mostly consists of estimates of false positives (ΩV). Increasing the sample size results in an increase in S at the cost of a decrease of V. Modification of this mixture explains the modification of the shape of the distribution of the distribution for the ΩR estimates.

Figure 4: Parameter estimates with varying n for all selected variables.
Each of the four panels was obtained with a specific sample size with n={100; 200; 400; 1000}. Whatever the panel, the following distributions were plotted: 1-distribution of the estimates for the ΩR sets over 200 identification sets parameters (histogram with horizontal hatching). 2-distribution of the estimates of the ΩR sets over 200*50 validation datasets (histogram with diagonal hatching). The vertical dotted line indicates the mean of the latter distribution. The vertical continuous line indicates 0.2.
The R selected variables are a mixture of False Positives (V) and True Positives (S) that respectively corresponds to the left and the right mode of the distributions of ΩR estimates. When increasing the sample size, S increases at the cost of a decrease of V, thus modifying the shape of the distribution of the estimates for the ΩR variables. When n=1000, there are fewer FP and the estimates for TP are no longer over-estimated. This shows the consequence of the regression to the mean phenomenon in terms of confirmation of the selected candidate biomarkers.
In real life studies, variables are selected according to the strength of association estimated during the identification step. The strength of association has then to be re-estimated during the validation step, in order to confirm the effect on survival of the corresponding candidate biomarkers. It appears that the distance between the estimates on the identification and the validation datasets is high with n=100 individuals: when re-estimating the strength of association of ΩR on independent datasets, it falls and tends toward 0. This divergence between the first estimation and the re-estimation is an illustration of optimism. Thus, it shows how regression to the mean leads to optimism. By quantifying this divergence, it is possible to quantify optimism. With n=1000, distributions of the estimates for Ωp0 and Ωp1 variables fluctuated to a lesser degree. Thus, selection bias and regression to the mean decreases; ΩR is almost completely composed of ΩS and the two distributions superimpose: there are fewer FP and the estimates for TP are no longer over-estimated.
These results demonstrate how a biased estimation of the parameters on the identification sets influences the selection of TP and FP, and illustrates how power increases and optimism decreases with increasing sample size.
These comments demonstrate why large sample sizes in highdimensional studies are important. Indeed, the estimation of the strength of association from the identification step is critical, because it influences regression to the mean through the selection of variables, and therefore, their validation on new independent datasets. This is important to keep in mind when calibrating new “omic” studies. At present, many current studies are designed to identify new markers on small sample sizes; this choice is justified by claiming that the candidate biomarkers will be validated on larger sample sizes. In complement to Figure 4, Figure 5 shows that this reasoning is incorrect (same legend as for Figure 4), and prevents the identification of relevant markers. The above panel of Figure 5 shows the results obtained with identification and validation datasets of respectively 100 and 1000 individuals. Because of the poor estimates obtained with n=100, many variables are wrongly selected, and have far lower estimates on validation datasets, due to regression to the mean. As demonstrated through the above results, the identification step can only be improved by generating non-biased estimates of the strength of association for Ωp; this is made possible by using larger sample sizes in the identification step. Increasing the size of validation sets cannot improve the first estimation obtained during the identification step. This is confirmed on the bottom panel of Figure 5, where estimation and selection were conducted on identification sets of 1000 patients and validated on 100 patients. This time, the strength of association is correctly estimated on the identification sets, and thus, confirmed on the validation sets, even though they are of smaller size.

Figure 5: Parameter estimates for all selected variables with n={100; 1000} and n={1000; 100}, respectively, for identification and validation sets. At the top n={100; 1000}; at the bottom n={1000; 100}, respectively, for identification and validation sets. The following distributions were plotted: 1-distribution of the estimates for the ΩR sets over 200 identification sets parameters (histogram with horizontal hatching). 2-distribution of the estimates for the ΩR sets over 200*50 validation datasets (histogram with diagonal hatching). The vertical dotted line indicates the mean of the latter distribution. The vertical continuous line indicates 0.2.
With n=100, estimates of the strength of association are poor and have far lower estimates on validation datasets, even large ones like with n=1000. With n=1000 in the identification datasets, however, the strength of association is correctly estimated; as a consequence, it is confirmed on the validation sets, whatever their size.
All results discussed above were obtained when the 20 most relevant variables were selected using the log-rank statistics. Similar results were obtained with control of the FDR (results not shown). In fact, controlling the proportion of FP does not correct the estimation and selection bias, thus also leading to regression to the mean and optimism. The main difference lies in the way variables are selected. When n is small, the estimation bias is high, therefore, many FP would be selected; thus type I correction leads to small R. When n increases, both R and S increase so that the proportion of FP stays constant. Thus, variables with lower strength of association than the simulated ones have to be selected to satisfy this constraint. For this, the minimum value of selected variables is moved to the left when n increases (results not shown), leading to a decrease of the selection bias.
Nevertheless, these results were observed when using a high value of FDR (FDR=0:2). This high value was chosen to mimic the exploration situation. It would be interesting to evaluate the influence of the choice of the FDR value on these results.
In this paper, we chose to focus on the regression to the mean phenomenon, to explain and illustrate through a simulation process how it takes a major place in the mechanisms of identification and validation of candidate biomarkers. However, it is useful to keep in mind that this is not the only issue that may affect the relevance of “omics” studies.
In particular, we chose not to consider in the simulation process, the role of the variables database defining the set wherein candidate markers will be searched for and selected. In fact, the selected candidates are highly dependent on the choice of the initial set of variables. In the case of genomics or transcriptomics, for example, this will influence the choice of the array. This choice is oriented by the clinical question underlying the study. In the case of clinical proteomics in discovery stage, the initial database reference is partially defined by the type of biological material used as sample (blood, tissue, biopsy, etc...), and by the retained purification method. Selected candidate biomarkers highly depend on these choices, as the reference database will highly vary depending on the type of purification, and/or biological material. It is to note that for clinical proteomics in discovery stage; by contrast with genomics or transcriptomics, the choice is only partial because the exact content of proteins or peptides under study is not known a priori. Moreover, only proteins already identified in known data banks will be used. These choices may then be analyzed in complementary ways to take benefit from the distinct information coming from each of them. In the case of proteomics dataset, another issue may occur. In fact, the reference database may not exactly reflect the content of the processed sample due to technical artifacts like limit of detection, and/or resolution of the measure instrument, statistical preprocessing, and so on. This leads to “technical” missing values that are then missed from the statistical study and this without any biological basis.
To sum up, the database reference is a finite ensemble, and is chosen with an a priori knowledge, and this a priori may lead the investigator to miss some interesting candidate biomarkers.
The objective of this work was to demonstrate how the discovery of important variables directly follows from the estimation of effect sizes, and how it is also influenced by the selection to the mean phenomenon. In fact, the two questions cannot be separated, as this of the statistical power. When searching for new markers, the true strength of association is not known. Sampling of the population concerned is used to obtain an unbiased estimation of it, and thus, to select relevant markers among a large number of “omic” variables. This exploratory stage involves two must-have steps: an identification step and a validation step. During the identification step, potential markers are selected on the basis of their estimated strength of association. In this work, we showed how the selection process influences regression to the mean. Understanding the phenomenon is a first step to overcoming the problems caused by regression to the mean. Only variables with extreme estimated strength of association are selected. This leads to a selection bias that is all the more strong that sample sizes are low. This favors regression to the mean and optimism, the impact of which is then highlighted through validation studies, with bad consequences. In fact, it will finally lead to the selection of variables wrongly considered as candidate biomarkers.
Pertinent “omic” clinical studies are only possible if the strength of association is estimated in a non-biased way in the identification step. Consistent sample sizes will have two effects: 1-improvement in the accuracy of estimates due to regression to the mean. 2-gain in power. Only then will it be possible to identify relevant markers whose effects will be confirmed on independent datasets, and thus be used in the clinical practice.
We wish to thank Philip Bastable for editing the manuscript.