Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2009) Volume 2, Issue 3
Phosphopeptide/protein identification using tandem mass spectrometry (MS/MS) is a challenging issue in proteomics research. In particular, phosphopeptides typically exhibit low intensity peaks of b and y ions in spectra when serine or threonine is phosphorylated. Consequently, the existing algorithms for peptide and protein identification generate a high false discovery rate when coping with phosphopeptide spectra. In order to increase the number of correct phosphopeptide identifications using database search, a new data mining approach for spectra preprocessing is proposed. A support vector machine classifier is used to calculate the probability of each peak representing a b or y ion. Next, low-probability peaks are removed from spectra, while remaining peaks have their intensities enhanced. As a result, a huge increase in signal-to-noise ratio is provided and the chances of detecting important peaks are significantly advanced. Experiments using MASCOT and SEQUEST along with Peptide/ProteinProphet and a decoy database approach showed a significant improvement in the sensitivity of phosphopeptide identification without compromising specificity, demonstrating that our new strategy for MS/MS spectra preprocessing is a powerful proteomics tool for enhancing phosphopeptide identifications.
Keywords: Data mining, Tandem mass spectrometry, Spectra preprocessing, Phosphoproteomics, Peptide/protein identification.
Protein phosphorylation is a key post-translational modification for many cellular processes in all living organisms (Morandell et al., 2006). Moreover, abnormal phosphorylation can be responsible for many serious diseases such as diabetes and cancer (Kocher et al., 2006). Therefore, a complete understanding of many biological processes and signaling pathways depends on a profound analysis of involved phosphorylated proteins.
Liquid chromatography (LC) coupled with tandem mass spectrometry (MS/MS) has been the method of choice for protein identification in complex mixtures (Steen and Mann, 2004). On the other hand, such a high throughput approach can generate thousands of MS/MS spectra in a single run, turning the manual interpretation into an infeasible practice (Kapp et al., 2005). By that means, computational tools for spectra interpretation became vital for large-scale MS-based proteomics. However, the most popular algorithms for peptide and protein identification produce a high false discovery rate (FDR) (Kapp et al., 2005). This is mainly due to the presence of background signals in spectra, i.e., peaks representing isotopes, Internal fragments, electronic noise, chemical noise, and ions originated from unknown fragmentation pathways (Gentzel et al., 2003; Mujezinovic et al., 2006). In the case of phosphopeptides, there is an additional source of complexity. Phosphopeptide fragmentation in low energy dissociation tends to occur in phosphate groups when serine or threonine is phosphorylated, which is normally the case. Hence, it is common to see a very prominent peak in the central region of the spectrum (Figure 1), corresponding to neutral losses of HPO3 or H3PO4 groups from the precursor ion (Kocher et al., 2006). Therefore, other necessary fragmentations to form b and y ions are scarce. The resulting low signals of such ions are then easily confounded with noise peaks (Figure 1). For this reason, false positive (FP) identifications occur even more frequently for phosphopeptide spectra interpretation.

Figure 1: Typical phosphopeptide MS/MS spectrum. The reported sequence indicates phosphorylation in the first and third serines. Notice the intensity of the central peak at m/z = 1040 compared to intensities of other peaks. This prominent peak corresponds to the neutral loss of two H3PO4 groups ( − 196 Da, offset of − 98).
In order to avoid misinterpretations, various algorithms have been proposed to process spectra before ion search. One approach focuses on the deconvolution of multiply charged peaks and deisotoping (Ferrige et al., 1991; Reinhold and Reinhold., 1992; Zhang and Marshall, 1998). More recently, other cleaning steps have been included. Gentzel et al., (2003), for example, considered peak centroiding, joining of redundant spectra and automatic calibration. However, no special procedure thereby was undertaken for noise elimination. Mujezinovic et al., (2006) propose a sophisticated noise removal procedure, but, as described by the authors, this approach is only robust for mild inaccuracies in spectra. The main idea in the work of Jaitly et al., (2004) was to elucidate peaks of interest instead of background peaks. A maximum likelihood estimate is used to classify peaks corresponding to fragment ions, achieving high sensitivity and specificity. Nevertheless, no experiment is presented in this work concerning the impact of peak classification on spectra interpretation. All cited studies provided an important enhancement on MS/MS spectra preprocessing methods. However, to the best of our knowledge, the particular issue of low quality of phosphopeptide spectra remains unaddressed. Recent works ( Ishihama et al., 2007;Imanishi et al., 2007; Lu et al., 2007; Hoffert et al., 2007have been proposed for a more efficient validation of phosphopeptide identifications. These methods act as good filters to separate correct from incorrect outcomes. Nonetheless, no effort is presented to improve the quality of phosphopeptide spectra in order to decrease the number of incorrect sequence assignments.
In this paper, we propose a new data mining approach for advancing phosphopeptide/protein identification in standard database (DB) search tools by introducing a novel preprocessing modality for spectra before ion search. The method trains a support vector machine classifier for determining the useful peaks in a spectrum concerning the procedure of assigning a peptide sequence. An important characteristic of the proposed method is that, instead of having an unique training set (TS), a particular TS is dynamically constructed for each dataset in analysis so that a specialized classifier can be obtained for each run. This step provides high flexibility concerning variations in spectra caused by different acquisition or sample preparation methods as well as the mass spectrometer type used. A 10-fold cross validation showed a classifier accuracy of around 80%. In our experiments, we verified that about 84% of peaks in the original spectra could be eliminated after preprocessing (after applying the learned classifier), yet the identifications could be improved. In a decoy DB analysis on SEQUEST (Eng et al., 1994) results, for instance, we could observe increases in the number of correct assignments varying from 33% to 60%. The same analysis on MASCOT (Perkins et al., 1999) results showed enhancements between 20% and 35% when employing the identity threshold. The statistical models provided by Peptide/ProteinProphet (Keller et al., 2002; Nesvizhskii et al., 2003) confirmed the power of our method. The sensitivity curves plotted for the preprocessed datasets dominated the sensitivity curves for original data in all cases. The error curves for treated spectra, on the other hand, were mostly dominated by the error curves of noisy data. Examining the proteins infered by ProteinProphet, a higher number of matches and better coverage could be normally observed after the application of our preprocessing procedures.
Sample Preparation and LC/MS
In two independent experiments, samples were prepared according to the following protocol. Cytosol of Mek1-/- MEFs was pretreated in a KESTREL-like approach (Knebel et al., 2001), followed by in vitro kinase assay using Chemical Genetics tools (Shah et al., 1997). Samples were incubated with or without an ATP-binding pocket mutant of constitutive active GST-Mek1 in the presence of a N6-modified ATP-analog. Proteins were precipitated according to Wessel and Flugge, (1984) and a proteolytic digestion with trypsin was performed as described by de la Fuente van Bentem et al. (2006). For the enrichment of phosphorylated peptides, the samples were methylester-modified with normal (light) or deuterated (heavy) methanol (Ficarro et al., 2002), and the IMAC protocol was performed (de la Fuente van Bentem et al., 2006) using two different elution steps with 50mM and 125mM Na2HPO4, respectively. In a third experiment, samples were prepared as above, except for the in vitro kinase assay
Eluted peptides were separated by reversed phase high performance liquid chromatography (RP-HPLC) coupled to a LTQ FT mass spectrometer (hybrid linear ion trap / Fourier transform ion cyclotron resonance (Thermo Electron, Bremen)) using a multistage activation method as described in Schroeder et al., (2004).
MS/MS Data
For each experiment described above, we took data files in Xcalibur raw format corresponding to the samples with GST-Mek1, methylester-modified with normal methanol, eluted with 125mM Na2HPO4. A total of three raw files were then converted to dta files, resulting in 24405 (SET1), 23668 (SET2) and 18996 (SET3) spectra, respectively. SEQUEST (Bioworks v3.3, Thermo Electron) was run on these three datasets so that we could filter high-quality identifications for constructing the TSs. In order to evaluate the preprocessing procedures, we randomly picked 10000 files (spectra) from each dataset above (excluding spectra used to build the TSs). After applying our approach on these selected spectra, three additional sets of spectra were generated, resulting in six datasets for the analysis described in Section "Results and Discussion", three of them containing original 10000 spectra each, and the others containing the respective preprocessed versions. These datasets are defined here simply as SET1_O, SET2_O, SET3_O, SET1_P, SET2_P and SET3_P, respectively, where “_O” and “_P” indicate original and preprocessed data.
In order to perform experiments using MASCOT (v2.2, Matrix Science), each set was converted into the MASCOT generic format mgf. All searches in MASCOT and SEQUEST were performed against the mouse IPI database (v3.18) (Kersey et al., 2004). The search parameters were set the same for all runs. Enzyme: trypsin; missed cleavages: up to 3; fixed modifications: carbamidomethyl (C), methyl (C-term), Methyl (DE); variable modifications: oxidation (M), phosphorylation (ST), phosphorylation (Y); peptide charges: +1, +2 and +3; protein mass: unrestricted; mass values: monoisotopic; peptide mass tolerance: ± 10 ppm; fragment mass tolerance: ± 0.6 Da.
Data Mining Framework for Spectra Preprocessing
Figure 2a illustrates the general overview of our new framework for spectra preprocessing. Initially, the original spectra are used for the TS construction (details in Figure 2b). Next, a support vector machine classifier is trained using the constructed TS. Finally, the resulting model is used to classify peaks and adjust intensities (details in Figure 2c) in the original spectra, giving rise to more easily interpretable spectra. The following sections provide detailed descriptions of each step.

Figure 2: Framework for spectra preprocessing. (a) General overview: The TS is constructed as illustrated in (b). The obtained TS is then used to train a support vector machine classifier. In the last stage, the resulting model is applied to the original spectra for classifying peaks and adjusting intensities. (b) Dynamic TS construction details: A standard DB search engine for peptide identification (SEQUEST in our case) is run a priori to provide spectra interpretations. Next, filters are applied to separate high-quality identifications. From the resulting filtered set, peaks are taken to form the first TS version. In order to optimize the classifier’s accuracy, three additional steps are performed: Balancing of TS, discretization and feature selection, producing a final TS for the model construction. (c) Pseudo-code for peak classification and intensity adjustment: The classifier is used to assign a probability P to each peak. Low-probability peaks are removed from spectra, while the remaining peaks have their intensities adjusted according to P.
Applying Filters for High-quality Identifications
In the proposed framework, TSs are constructed on-the-fly, i.e., one for each dataset in analysis. In order to obtain peaks for representing instances in the TS, a set of MS/MS spectra with respective correct sequences is necessary. For this purpose, some standard database search tool is run a priori on the data. In our experiments, SEQUEST was chosen to provide interpretations of spectra. Next, strict filter constraints are applied on the obtained identifications for minimizing the risk of selecting incorrect sequence assignments (for minimizing noise in the TS). These constraints are as follows:
The application of the described constraints leads to a very reduced set of spectra. The idea is to achieve a high precision, avoiding noise in the TS. Experiments demonstrated that about 50 spectra are enough to provide a TS containing more than 1000 peaks (TSs can be found in supplementary data - appendix "Addendum: Web Supplement"). We assume that peaks have similar patterns among spectra in the dataset. Therefore, even learning with a reduced subset, we would be able to accurately classify peaks in the remaining spectra. The validity of this assumption is demonstrated in our statistical evaluations described in Section "Statistical Analysis", since the identifications for preprocessed spectra are improved. After applying the constraints, if the number of selected spectra is not enough to provide a significant TS, we suggest changing the percentage in item vi, as itens iv and vii already provide strong cutoffs. It is also a good alternative to test changing the minimum ΔCn (item iv) or even the percentage of b and y ions found (item vii) until getting a combination of values leading to a suitable TS. However, the values stated above are normally apropriate. In the case of SET3, for instance, only item vi was altered, i.e., we selected the best 20% results to obtain enough spectra. For the other datasets, the restrictions were applied exactly as proposed. A total of 68, 49 and 47 spectra were selected from SET1, SET2 and SET3, respectively, to form each training set.
Selecting Peaks
Once having selected spectra and their assigned sequences the next step is to separate peaks in spectra to form the TS. For each spectrum, peaks are labeled golden if they match to the theoretical b and y m/z values (item vii), but only ions of charges +1 and +2 are considered. Any other peak is labeled as background. In this manner, signals representing internal fragments, isotopes, electronic and chemical noise, the precursor and its derivatives, neutral losses, and b/y ions of higher charges are taken as background. Our assumption is that leaving only peaks representing b+1/+2 and y+1/+2 ions in spectra is the cleanest and simplest way to minimize misinterpretations. Besides, the m/z values for ions of higher charges are difficult to handle due to resolution issues. After separating golden and background peaks, each one has its features extracted from spectra (Section "Feature Selection") and is used to compose the first TS version.
Balancing of TS
In many real-world classification problems, the majority of instances in a TS is from one of the classes. In binary classification, the minority class is normally the class of interest. This situation occurs in our case. The first TS version obtained from SET1, for example, contained 40184 peaks of the class “background” and 1022 peaks of the class “golden”. Such an imbalanced TS often causes machine learning algorithms to perform poorly on the minority class, i.e., the rare instances are often treated as noise (Dehmeshki et al., 2003). In order to make both classes have the same weight for the model construction, the TS is balanced using a supervised instance resampling algorithm. This algorithm produces a random subsample of instances in the class background, applying a sampling with replacement approach (Hsia, 2005), resulting in the same number of examples in both classes. In this stage, the second TS version is established.
Feature Discretization
The third TS variant is generated from a discretization of the given numeric features (Section "Feature Selection"). According to Witten and Frank, (2005), learning methods that are able to handle numeric features often produce better results and work faster when features are discretized beforehand. Experiments confirmed that the classification accuracy is improved after discretization (supplementary data). In our approach, we employ a supervised discretization algorithm that uses an entropy minimization heuristic for discretizing the range of a continuous-valued feature into multiple intervals (Fayyad and Irani, 1993; Witten and Frank, 2005).
Feature Selection
In order to accomplish our classification, we had to select important features that clearly distinguish golden peaks from background signals. For the employed mass spectrometer (Section "Sample Preparation and LC/MS"), peaks of b and y ions are normally accompanied by signals with lower m/z values representing neutral losses such as water, ammonia, two molecules of water, and water+ammonia (Frank and Pevzner, 2005). The occurrence of a ions is common as well and these ions can undergo a neutral loss of water or ammonia (Frank and Pevzner, 2005). Isotope variants (more commonly +1 Da and +2 Da) are also very frequent and can present the same cited satellite peaks. Putting together all these information and initially considering +1 charged ions, the first set of features of a given target (peak being classified) had its composition based on the presence of the described satellite peaks at the positions shown in Figure 3a. However, in order to increase the classifier’s discriminatory power, we added features regarding also background peaks, as demonstrated in Figures 3b and 3c. If we consider a signal representing loss of water as the reference, for example, satellite peaks are expected to be found at the positions shown in Figure 3b. Hence, it is appropriate to include in the set of features also the occurrence of peaks at the positions - 25,- 10, - 9, - 8, +3, +18, +19 and +20 in order to improve the distinction between signals representing loss of water and golden peaks. On the other hand, when taking as reference a peak representing a loss of CO (a ion), its expected vicinity is composed as stated in Figure 3c. Consequently, the inclusion of the positions - 8, - 7, - 6, - 5, +10, +11, +12, +13, +28, +29, +30 in the set of features increases the dissimilarities between peaks representing a ions and golden peaks. This reasoning was applied for the 17 satellite peaks shown in figure 3a, giving rise to 76 distinct features. Since +2 charged ions are also considered, peak positions are divided by two, ending up with 128 features (the division leads to many repeated positions). Finally, since noise can be very abundant in spectra, random signals might occupy the described positions, being thus confounded with satellite peaks. As a result, instead of considering the occurrence of peaks at the proposed positions, the set of features was formed by the intensities of such peaks normalized to (divided by) the target’s intensity. Therefore, we always start with 128 features (128 normalized intensities of satellite peaks) for each dataset being operated. However, in order to remove irrelevant features, the final TS is generated using a correlation- based feature selection heuristic (Plant et al., 2006). This method assigns higher scores to feature subsets whose features show high correlation with the class and low correlation among each other.

Figure 3: Simplified scheme representing the vicinity of common peaks in spectrum for +1 charged ions. The peaks are shown in relative positions, indicating offset in Dalton. (a) Golden peak (red) and peaks appearing frequently in its vicinity (blue). (b) Changing the reference to a peak representing loss of water (red). (c) Changing the reference to a peak representing loss of CO (red).
Learning with Support Vector Machines (SVMs)
SVMs (Cristianini and Shawe-Taylor, 2000) constitute a very powerful classification method with outstanding performance applied to many biomedical problems (Noble, 2006). Given instances xi, i = 1, ..., l with labels yi∈{1, -1}, the training of SVMs is the task of solving the following quadratic optimization problem:
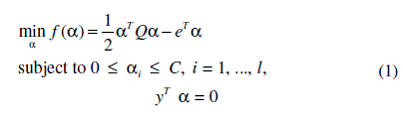
where e is the vector of all ones, C is the upper bound of all variables, Q is an l×l symmetric matrix with Qij = yiyj K(xi, xj), K(xi, xj) is the kernel function, and the optimal weights αi are searched for defining a hyperplane that separates the two classes so that unknown instances can be accurately classified. In our case, a pair (xi, yi) represents a peak in the training set, where xi is the vector of features (normalized/discretized intensities) and yi denotes whether the peak is golden (1) or background (-1).
The size of the matrix Q is proportional to the square of the number of training examples, which can lead to huge storage requirements. Consequently, decomposition methods are applied to modify only a subset of the vector α in each iteration. In this context, we chose a powerful method termed LIBSVM (Fan et al., 2005) to implement in our framework, as this algorithm presents faster convergence compared to previously proposed approaches, without compromising the result quality. Another advantage of LIBSVM is that it is able to provide a probability estimate to determine the confidence level of a classification instead of predicting “hard” class labels (Wu et al., 2004). This is very suitable to our goals, as can be seen in the next section.
We selected the radial basis function (Cristianini and Shawe-Taylor, 2000) as the kernel function with γ = 1/k, where k is the number of features. Using 10-fold cross validation, the performance of LIBSVM for golden peaks classification in the final TSs gathered from SET1, SET2 and SET3 is summarized in Table 1.
| Data set | Sensitivity | Specificity | Accuracy | AUC |
|---|---|---|---|---|
| SET1 | 75.4 | 84.1 | 80.58 | 0.873 |
| SET2 | 75 | 80 | 78 | 0.835 |
| SET3 | 77.6 | 81.6 | 80.05 | 0.863 |
AUC is area under the ROC curve.
Table 1: Performance of LIBSVM for golden peaks classification using 10-fold cross validation in the final TSs built from SET1, SET2 and SET3.
Classifying Peaks and Adjusting Intensities
The SVM classifier is used in the proposed framework to calculate the probability P of a peak designated to be golden. The peak is only kept in spectrum if P> 0.5. In this case, its intensity is set to P ×100 (algorithm in Figure 2c). As a consequence, only the most significant peaks for peptide sequence identification are maintained in order to avoid FP matches caused by additional mass combinations of background peaks. The adjustment of intensities is also important to decrease the number of wrong answers, since many popular tools for peptide and protein identification consider only the most intense peaks in the ion search procedure. The SVM runs took on average 1.02ms, 0.49ms and 0.37ms (the resulting TS from SET1 had a higher number of features) per peak for generating the datasets SET1_P, SET2_P and SET3_P, respectively.
In this paper we applied a new data preprocessing method for MS/MS spectra with the objective of enhancing the rate of correct phosphopeptide/protein identifications in standard DB search tools. Experiments using the proposed approach were performed on three selected datasets (SET1-3) generated from phospho-enriched samples, with results achieving a significant positive effect on the quality of phosphopeptide assignments. Since no reference data was available during this work, the experiments were based on a comparative analysis between results before and after spectra preprocessing.
First, a manual inspection of MASCOT and SEQUEST results was performed. An important observation was the huge increase in signal-to-noise ratio (S/N) for treated spectra as illustrated in Figure 4 (another example can be seen in Figure 2S of supplementary data). Moreover, in cases in which original and preprocessed spectra led to the same peptide sequence, a higher number of ions in the preprocessed spectra was frequently observed (Figure 4 and Figure5). Even in cases in which fewer ions were found, higher scores were normally generated as a consequence of a better S/N and intensity adjustment.

Figure 4: Comparison of MASCOT results for a spectrum before and after preprocessing, supporting the same peptide sequence HNQDSQHCSLSGDEEDELFK. (a) Original spectrum (SET1_O). (b) Preprocessed spectrum (SET1_P). (c) Detail of original spectrum (m/z: 350 to 450). (d) Detail of preprocessed spectrum (m/z: 350 to 450). In (b), it can be seen a huge decrease in the number of peaks, yet a higher number of ions was found. In (a) only 3.98% of peaks are annotated, while in (b) this value raised to 55.05%. The treated spectrum shows that MASCOT was able to detect 7 new golden peaks (in blue) that were hidden in the background before preprocessing. Nevertheless, b(15) got lost. In (c) and (d) the efficiency of the cleaning and the intensity adjustment steps for the zoomed range is demonstrated (another range can be seen in Figure 1S of supplementary data). See supplementary data for the complete result report and for zooming other regions of the figure.

Figure 5: Comparison of SEQUEST results for a protein identified before and after preprocessing. (a) SET1 O. (b) SET1 P. In (b), three new sequences were included, resulting in a higher number of matches and better coverage. Comparing spectra with the same scan number in (a) and (b), the quality values were clearly more favorable after preprocessing. The symbol“#” means phosphorylation, while “*” denotes oxidation of methionine.
We have observed that peaks representing neutral losses of water and ammonia were regularly kept in spectra. It is probably due to the fact that these ions have similar properties when compared to b and y ions, such as the presence of isotopes and further water losses, suggesting similarities in their vicinities as well. However, since standard DB search algorithms expect the occurrence of water and ammonia losses, such misclassifications did not prevent our method from yielding to important improvements (Section "Statistical Analysis"). Reviewing Figure 4b, we can also observe some nonannotated peaks. This is either due to their lower intensities (i.e., they have P close to 0.5) or due to the fact that they represent ions (precursor and its derivatives, internal fragments, etc) with properties similar to golden peaks, alike water and ammonia losses. But, even with some misclassifications, the increase in S/N was remarkable.
Another kind of manual inspection we performed was the analysis of the number of matches and the coverage for the same proteins identified before and after preprocessing. This investigation strongly indicated that many spectra could be correctly interpreted after preprocessing. An example from SEQUEST results is shown in Figure 5. For the given protein identified in SET1_O and SET1_P, besides observing a higher number of matches and better coverage for the second dataset, we can also notice that the quality parameters were mostly better for the peptides reported in the preprocessed case, turning this protein into a significant identification with a much better position in SEQUEST ranking for SET1_P. For an extended manual inspection of the peptides and proteins identified in common between original and preprocessed data, see the complete result report of MASCOT and SEQUEST for all datasets in the supplementary data.
Statistical Analysis
In order to provide a comprehensive analysis of the overall benefit of our method, statistical measurements from SEQUEST, MASCOT and TransProteomic Pipeline (Keller et al., 2005) were considered. In a first analysis, we estimated the number of correct assignments made by MASCOT and SEQUEST. This calculation was performed using a decoy DB approach (Elias et al., 2005; Balgley et al., 2007). In this method, the search is repeated against a database with randomized or reversed sequences (decoy DB). As a consequence of this search, the number of positive matches can be used to estimate the FDR. MASCOT employs a decoy DB composed by randomized sequences. Here, the FDR calculation is based on the number of significant hits in normal and decoy DBs. Note that, in MASCOT, a peptide identification is regarded as significant if its score is above the homology or identity thresholds, the latter being the strongest quality measure (Perkins et al., 1999). Table 2 shows the comparative analysis after MASCOT search where a significantly improved sensitivity (= rate of correct hits obtained) and specificity (= rate of incorrect hits below the employed threshold) could be in general demonstrated. When using the identity threshold, the number of estimated correct hits increased 20% (from 175 to 210 ), 35% (from 48 to 65) and 33% (from 94 to 125), respectively, after spectra preprocessing. For the homology threshold, though in a lower percentage, all treated datasets presented improvements in the number of correct hits as well. Note yet that, except for one case (from 3.7% to 4.46%), the FDR was systematically diminished.
| # of hits in Mouse DB | # of hits in Decoy DB | FDR | |
| SET1_O | 176 | 1 | 0.57 % |
| 378 | 14 | 3.7 % | |
| SET1_P | 210 | 0 | 0 % |
| 404 | 18 | 4.46 % | |
| SET2_O | 48 | 0 | 0 % |
| 170 | 8 | 4.71 % | |
| SET2_P | 65 | 0 | 0 % |
| 186 | 7 | 3.76 % | |
| SET3_O | 99 | 5 | 5.05 % |
| 288 | 16 | 5.56 % | |
| SET3_P | 126 | 1 | 0.79 % |
| 305 | 10 | 3.28 % |
Table 2: Analysis of significant hits in MASCOT results using a decoy DB approach. In the first line of each case the values are based on the identity threshold, while in the second line the homology threshold is the reference.
Since SEQUEST does not provide any significance threshold for peptide identifications, we employed several score thresholds for calculating the FDR. It should be noted that such experiments have to be done separately for each result because a particular threshold used in a dataset does not necessarily provide the same FDR in another dataset. For our experiments, a decoy DB was generated by reversing the peptide sequences contained in the original DB (Elias et al., 2005; Balgley et al., 2007). A broad range of Xcorr thresholds was used for each dataset in order to obtain FDRs varying from 0 to 0.2. Figure 6 shows for the same FDRs that preprocessed datasets lead normally to a higher number of correct assignments. Even having some regions in Figure 6b and 6c in which the curves coincide, it is clear the dominance of the blue curves in all cases. For some FDR intervals, it can be seen an augmentation in the number of correct identifications of up to 40%, 60%, 33% in the datasets, respectively, after using our approach.

Figure 6: Decoy DB analysis for SEQUEST results. (a) SET1_O/P. (b) SET2_O/P. (c) SET3_O/P. Each data point corresponds to a different Xcorr threshold. The x axis indicates the estimated FDR (calculated by counting the hits above the threshold for the decoy DB), while the y axis shows the number of hits above the threshold when using the mouse DB. The red curves correspond to the original datasets and the blue curves relate to the treated datasets. Taking the same FDRs in original and preprocessed data, a higher number of hits comming from the mouse DB was in general observed for treated spectra.
To further verify our studies, we used also Trans-Proteomic Pipeline (v3.2). This tool is a pipeline for analysis of LC-MS/MS proteomics data, containing modules for validation of peptide identifications (PeptideProphet) and protein inference (ProteinProphet). PeptideProphet was used to evaluate MASCOT and SEQUEST results. PeptideProphet uses the expectation maximization algorithm to compute probabilities of peptide assignments. Applying its statistical model, PeptideProphet provides the sensitivity/FDR tradeoff as ilustrated in Figure 7. The plots show that preprocessed datasets presented a better sensitivity, yet same or lower error in general for both MASCOT and SEQUEST when compared to the original data. Particularly in Figure 7b, the difference for treated spectra is very prominent.

Figure 7: Sensitivity and FDR curves provided by PeptideProphet. (a-c) SET1_O/P, SET2_O/P, and SET3_O/P, respectively, using MASCOT. (d-f) SET1_O/P, SET2_O/P, and SET3_O/P, respectively, using SEQUEST. The x axis shows the different probability thresholds, while the y axis designates sensitivity and error. The blue and the red curves represent sensitivity and FDR, respectively, of untreated data, while the black and the green dashed curves describe sensitivity and FDR, respectively, of preprocessed spectra. The dominance tendency of the black curves upon the blue ones as well as the red curves upon the green ones indicates that the sensitivity was improved, yet the error decreased in the datasets obtained after application of the proposed method.
In Figure 8, Venn Diagrams are shown. The diagrams contain the number of identifications reported by PeptideProphet with probability 0.9 or higher. This threshold is appropriate as the error rates for this value were low (varied from 1% to 3%. See supplementary data). It can be noted that the number of exclusive identifications for preprocessed data was always higher than for initial datasets.

Figure 8: Venn Diagrams of peptide identifications reported by PeptideProphet with Prob ≥ 0.9. (a) Using MASCOT results. (b) Using SEQUEST results. The number of exclusive identifications is always higher in treated datasets for both tools.
In Figure 9, our analyses are moved to the protein level by means of ProteinProphet. This program uses PeptideProphet results to identify proteins and to associate probabilities to these identifications. As can be seen in Figure 9, ProteinProphet provides also the sensitivity and FDR curves, which open again the possibility to compare the different scenarios for the tested datasets. The Figure reveals once more the improvements in sensitivity and specificity for the datasets produced by our method. In the protein level, the enhancements are even more obvious. The area between black and green curves (related to treated data) is clearly higher than the area between blue and red curves (regarding the unprocessed data) for all cases in both MASCOT and SEQUEST runs.

Figure 9: Sensitivity and FDR curves provided by ProteinProphet. (a-c) SET1_O/P, SET2_O/P, and SET3_O/P, respectively, using MASCOT. (d-f) SET1_O/P, SET2_O/P, and SET3_O/P, respectively, using SEQUEST. The description of these plots is the same provided in Figure 7, with the difference that here the analysis is in the protein level. Once more the preprocessed datasets demonstrated a better sensitivity and specificity (lower FDR). The enhancements in the protein level are even more prominent than in the peptide level (Figure 7).
Finally, ProteinProphet was used also to compare the same proteins identified in original and preprocessed data. For each protein identified in both cases, we computed the number of peptide matches and sequence coverage in order to verify if both measurements get improved after preprocessing. Figure 10 shows this comparison. It can be seen that the occurrence of proteins with more matches or higher coverage is much more frequent in treated than in original datasets.

Figure 10: Analysis of ProteinProphet concerning the number of proteins with more matches / better coverage among the same proteins identified before and after the application of the proposed method. The red region contains the number of proteins having more matches/higher coverage in original dataset. The blue region shows the number of proteins having more matches/higher coverage in preprocessed dataset. Finally, the yellow part holds the number of proteins in which no difference arose between data before and after preprocessing (the number of matches or the coverage did not change). In each comparison, the left bar shows the analysis related to the number of matches, while the right bar regards the coverage. The number of proteins with more matches or better coverage is normally much higher after preprocessing.
Storage and Runtime Requirements
The experiments described in previous sections were performed using a stand-alone PC: Pentium(R) 4 CPU 3.20GHz, 1GB RAM, under Windows XP. A significant decrease in storage requirements was achieved, as the number of peaks in treated spectra could be reduced on average by 84%. The running times of MASCOT and SEQUEST for preprocessed data, in turn, could be shortened by approximately 28%. However, additional processing time is necessary for TS construction and dataset treatment (Section "Classifying Peaks and Adjusting Intensities"). On the other hand, it should be noted that curators take days or even weeks for separating correct from incorrect identifications manually, which means that the additional time caused by our data mining method is therefore negligible. Furthermore, the overall processing time can be significantly reduced using a powerful computer cluster.
Existing computational tools for MS/MS spectra interpretation produce a high number of FP identifications. This is due to the presence of background signals in spectra, which introduces multiple mass combinations. Phosphopeptide spectra, produced by low energy dissociation, demonstrate an additional source of complexity because the phosphopeptide fragmentation is frequently poor, giving rise to low intensity peaks of b and y ions. Consequently, phosphopeptide spectra interpretation presents an even higher false discovery rate.
In order to decrease the number of FP identifications for phosphopeptides, we have developed a new data mining approach for spectra preprocessing. With a dynamic TS construction, our method trains a SVM algorithm to classify (cleaning step) and adjust intensities of peaks in MS/MS spectra. The cleaning step facilitates an elimination of up to 84% of peaks, leading to a huge increase in S/N, while the intensity adjustment step improves the chances of detecting important signals. Since we could not find any previously proposed methods for phosphopeptide spectra preprocessing, no comparisons could be performed between our procedures and other approaches. Nonetheless, the presented statistical evaluations demonstrate that our method improves sensitivity of phosphopeptide identification significantly without compromising specificity. A decoy DB analysis using MASCOT and SEQUEST searches showed that the number of correct hits could be significantly enhanced in all preprocessed datasets. Likewise, Peptide/ProteinProphet confirmed this benefit for both tools. In summary, our experiments demonstrate that our data mining framework for preprocessing MS/MS spectra is a powerful tool for enhancing phosphopeptide/protein identifications in standard DB search tools.
This work is supported by the Austrian Genome Program (GEN-AU), project Bioinformatics Integration Network (BIN II). The work in the Huber laboratory is supported by the Austrian Proteomics Platform (APP) within the GENAU, Vienna, Austria.
Addendum: Web Supplement
Supplementary resources are available at the website:
http://biomed.umit.at/page.cfm?pageid=515. It contains supplementary figures; SEQUEST, MASCOT and Trans- Proteomic Pipeline results; and training sets. The software is available on request from the authors.