Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Research Article - (2013) Volume 2, Issue 1
Keywords: Drug design, Aurora kinase, Virtual screening, Cheminformatics
Aurora kinases belong to serine/threonine kinase family and play important roles in cell cycle [1]. In this context, Aurora kinases have emerged as important drug targets since they play a major role in regulating cytokinesis during mitosis [2]. Mitosis is a vital process required for tissue regeneration, genomic development and to maintain the functional integrity of a cell [3]. So far, three types of Aurora kinases are reported in mammals, designated as A, B and C, which share a high sequence homology at amino acid level. In general, all Aurora family members are comprised of N-terminal domain, a catalytic domain and a short C-terminal domain [4]. Despite the presence of high conservation in domain architecture, Aurora proteins exhibit a high range of functional diversity [5]. For example, Aurora A (polar kinase) is primarily associated with centrosome separation [6,7]; Aurora B (equatorial kinase) is a chromosomal passenger protein [8]; while Aurora C appears to function in the centrosome from anaphase to telophase and is mainly expressed in testis.
Typically, kinase paralogs influence by regulating the cell cycle and their malfunction may lead to cancer phenotype. Accumulating evidence support the involvement of Aurora proteins in a wide range of tumors including breast [9,10], colon [11–13], pancreas [14], ovary [15], stomach [16], thyroid [17], head and neck [18]. This has increased the possibility of developing new anti-cancer drugs that could target Aurora kinases. Among these inhibitors, AT9283, AZD1152, PHA- 739358, MLN8054, MK-0457 and ZM447439 are of interest with specificities to type of Aurora kinases and are in clinical trials [19].
In this study, Virtual Screening (VS) was performed through Aurora focused library using Enamine database (www.enamine.net). Selected hits were subsequently used for developing three dimensional pharmacophore model which was then used for screening Princeton (www.princetonbio.com) and Uorsy databases (www.ukrorgsynth. com). Afterwards, retrieved hits were verified by molecular docking studies in order to gain insights into the structural features of these inhibitors and their binding analysis with Aurora kinase binding sites.
3D structure prediction
In the absence of a well-defined structure, homology modeling provides a rational alternative to develop a reasonable 3D model. Notably, homology modeling is currently the most accurate method for 3D structure prediction yielding models suitable for a wide spectrum of applications, such as investigations into mechanism, structurebased drug development and VS [20–23]. This approach can produce a reasonable structural model for any given protein sequence that has related templates having more than 25% amino acid sequence identity [24].
In order to elucidate 3D structures for human Aurora B and C paralogs, models bearing PDB IDs 2VGP and 2NP8 were used as templates, respectively. Swiss-Model server [25] was used for structure prediction followed by model optimization and validation using Ramachandran plot [26], Procheck [26], Errat [27], Verify3D [28] and WhatIF [29] tools.
Virtual screening
VS is usually described as a cascade of filter approaches to narrow down a set of compounds to be tested for biological activity against the intended drug target. Starting with a fast evaluation of druglikeness of compounds, VS is often followed by ligand-based and/or structure-based approaches, only if the target structure is available [30]. The computational methods employed in VS can be divided into three classes, with increasing complexity and computational requirements. In practice they are often used in combination. These include, evaluation based on two-dimensional (2D) property profiles [31]; evaluation based on a target-specific pharmacophore, which is a reduced representation of the key features in the target system or ligand [32]; and evaluation based on detailed 3D structure modeling of receptor-ligand interaction.
In this study, Aurora kinase focused library was taken from enamine database and used for VS. Enamine focused Aurora kinase library comprised over 2,575 compounds. The selected compounds were further evaluated using Aurora A as representative kinase by mobility shift assay. 2D property profiles were generated to check whether a compound is “drug-like” and whether it is synthetically accessible or orally bio-available. A simplest way of verification is the “rule of five” [33]. Compounds that fulfilled the “drug-like” criteria were selected for VS. The most relevant and detailed VS methodology involves objective docking of 3D models of both ligand and receptor targets. The quality of the fit between a given ligand and target is used to rank the ligand, or to predict the actual binding affinity by calibration with the known ligand-receptor complexes. The molecular docking approach requires 3D models of both target and compound library [34]. Compounds short-listed on the basis of binding energies were subsequently checked for toxicity, solubility, mutagenicity and tumorgenic effects by OSIRIS Property Explorer [35]. Hits showing high risks of undesired effects like mutagenicity or a poor intestinal absorption were eliminated, while compounds exhibiting low toxicity values were further selected.
Molecular docking studies
Automated dockings were performed on Aurora kinase focused library to locate the appropriate binding orientations and conformations of various inhibitors in the Aurora kinase binding pocket using AutoDock4.2 [36] tool according to the specified instructions. In brief, polar hydrogen atoms and Kollman charges were assigned to the receptor proteins. For ligands, Gasteiger partial charges were designated and non-polar hydrogen atoms were merged. All torsions for ligands were allowed to rotate during docking procedure. The program AutoGrid was used to generate the grid maps. Each grid was centered at the structure of the corresponding receptor. The grid dimensions were 80×80×80 Å with points separated by 0.375 Å. For all ligands, random starting positions, random orientations and torsions were used. The translation, quaternion and torsion steps were taken from default values indicated in AutoDock. The Lamarckian genetic algorithm method was used for minimization using default parameters. The standard docking protocol for rigid and flexible ligand docking consisted of 50 runs, using an initial population of 150 randomly placed individuals, with 2.5×106 energy evaluations, a maximum number of 27000 iterations, mutation rate of 0.02, crossover rate of 0.80 and an elitism value of 1. Cluster analysis was performed on the docked results using an RMS tolerance of 1.0 Å. The binding energy of each cluster is the mean binding energy of all the conformations present within the cluster; the cluster with lowest binding energy and higher number of conformations within it was selected as the docked pose of that particular ligand.
Molecular dynamic simulations
The hits obtained as a result of molecular docking were then subjected to molecular dynamics simulation studies. Due to the presence of conserved binding sites among Aurora kinases, we performed MD simulations using Aurora, a structure to study the behavior of drug-protein interactions. All MD simulations presented in this study were performed using Gromacs 4.5.5 simulation package [37,38] and the GROMOS96 43a1 force field [39], with the SPC water model [40]. The topology file and other drug specific force field parameters were generated using PRODRG tool [41]. The starting structures for Aurora A specific drug complexes were extracted through the docking experiments. Later, the Aurora A complexes with four different compounds which were prepared and subjected to MD simulations. The water box parameters were kept similar for all four complexes. These complexes were solvated in a dodecahedron box of dimension 1.0. Particle Mesh Ewald [42] was used to calculate the long-range electrostatic interactions and periodic boundary conditions were applied in all directions. All complexes were minimized first by the grompp module in GROMACS 4.5.5. As an appropriate number of counter ions are needed to neutralize the system. Our system needed one Cl- ion to make it neutralize. It was subjected to a steepest descent energy minimization until a tolerance of 1000 kJ/mol reached, in order to get rid of high energy interactions and steric clashes. The energy minimized system was treated for 100 ps equilibration run. Finally, the pre-equilibrated system was consequently subjected to 20 ns MD simulation run, with a time-step of 2 fs at constant temperature (300 K), pressure (1 atm) and without any position restraints [40]. Snapshots were collected every 10 ps and all analyses of the MD simulation were carried out by GROMACS analysis tools which includes g_rms, g_rmsd and g_gyrate. Based on the RMSD and RMSF analysis, simulations for 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid was extended further upto about 45 ns.
3D pharmacophore generation
A pharmacophore is defined as an ensemble of universal chemical features (hydrogen bonds, charge interactions and hydrophobic areas) that characterizes a specific mode of action of a ligand in the active site of the macromolecule in 3D space. This pharmacophoric pattern is the condition for ligand-macromolecule interaction. Searching these chemical patterns in large molecule databases allows the finding of new scaffolds for developing lead structures.
The compounds filtered on the basis of binding affinities to the potential ATP binding site of Aurora kinases [37], were subsequently ranked and grouped on the basis of their specificity, selectivity and affinity properties. Moreover, families were identified for selected compounds which included quinazoline, carboxamide and benzoic acid. Later, using these families, one representative from each family was selected on the basis of energy values and used for 3D pharmacophore generation by Ligand Scout tool [43]. The test set compounds included in this study were: (a) 2-(thiophen-2-yl) quinazoline; (b) N-[(E)- phenylmethylidene]-1H-imidazole-1-carboxamide; (c) 2-{[(1E)- (2-hydroxyphenyl)methylidene]amino}benzoic acid and a known inhibitor (d) 2-(1H-pyrazol-5-yl)-1H-benzimidazole (Figures 1a-1d).
Figure 1: 2D chemical structures of compounds isolated from Aurora focused library taken from Enamine. (a) 2-(thiophen-2-yl) quinazoline, (b) N-[(E)-phenylmethylidene]-1H-imidazole-1-carboxamide, (c) 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino} benzoic acid and (d) 2-(1H-pyrazol-5-yl)-1H-benzimidazole.
The best pharmacophore hypothesis was then used as a 3D query for screening Princeton and Uorsy databases. VS was implemented to retrieve the compounds from scaffold databases that fit the chemical features present in the best hypothesis. The screened hits were further filtered by applying Lipinski’s rule of five and ADMET properties. The compounds that satisfied Lipinski’s rule of five and ADMET properties were selected for molecular docking studies in order to determine the suitable orientation of leads and binding affinities with Aurora kinase binding sites.
Prediction of human Aurora B and C 3D structures
In order to predict the human Aurora B structure, crystal structure of Xenopus laevis Aurora B (PDB ID: 2VGP; resolution 1.70 Å) was obtained from PDB and used as template. MSA analysis indicated 80% sequence identity between target and template sequences and RMSD score was found to be 0.055Å (Figures 2a-2c). Aurora C model prediction was carried out using human Aurora A (PDBID: 2NP8, resolution 2.25 Å) as best template, which exhibited a sequence identity of 77%. The backbone RMSD between the 2NP8 and Aurora C modeled structures was 0.079Å, indicating a high structural identity (Figures 2d-2f). Ramachandran’s plot indicated that 99.1% and 99.3% residues lie in allowed regions for both Aurora B and C models, respectively. None of the active site residues were present in the disallowed region. Moreover, parameters like peptide bond planarity, non-bonded interactions, Cα tetrahedral distortion, main chain H-bond energy and overall G factor for both the structures lie within favorable range.
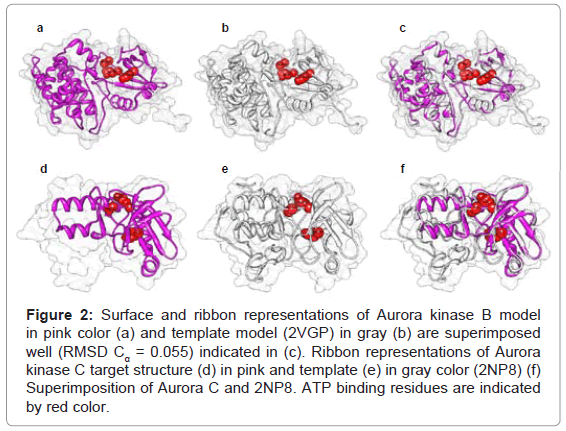
Figure 2: Surface and ribbon representations of Aurora kinase B model in pink color (a) and template model (2VGP) in gray (b) are superimposed well (RMSD Cα = 0.055) indicated in (c). Ribbon representations of Aurora kinase C target structure (d) in pink and template (e) in gray color (2NP8) (f) Superimposition of Aurora C and 2NP8. ATP binding residues are indicated by red color.
The homology models were further verified using Errat, Verify3D and WhatIF tools. Errat measures the overall quality factor for nonbonded atomic interactions and an accepted range of above 50 is considered for a high quality model. In our case, Errat scores were 95.2 and 87.7 for Aurora B and C, respectively. Verify3D employs energetic and empirical methods to produce averaged data points for each residue to evaluate the quality of structures. A score above 0.2 for more than 80% residues suggests a considerable high model quality. In both Aurora B and C, 92.19% and 98.73% residues exhibited score above 0.2. WhatIF was used to check the normality of local environment for amino acids. In this evaluation, the quality atomic distribution is determined around amino fragments. For a reliable structure, WhatIF packing scores should be above −5.0. In case of Aurora B and C models, none of the scores for any residue was found less than −5.0. These data indicated that Aurora B and C models are of good quality to allow further study.
Previously, at the ATP-binding pocket of Aurora kinase A, Leu139, Val147, Lys162, Glu211 and Ala213 residues were reported to be crucial for the potent ligand binding and kinase selectivity [44,45]. In case of Aurora B protein, Leu83, Val91, Lys106, Glu155 and Ala157 were found to be critical amino acids with respect to interaction [46]. Moreover, these residues were found to be highly conserved structurally among Aurora proteins. For identifying ATP binding pocket of Aurora kinase C, an alignment between Aurora A and C was carried out, which indicated that for Aurora kinase C, Leu49, Val57, Lys72, Glu121 and Ala123 residues were the critical amino acids for binding (Figure 2).
Molecular docking of selected inhibitors
The predicted Aurora structures were used as receptors to screen novel inhibitors. Protein-ligand docking was performed using AutoDock 4.2 [35] and each docked pose was analyzed individually to monitor the interactions on the basis of associated binding and docking energy values. Binding energy is the sum of intermolecular energy and torsional free-energy, while docking energy is the sum of intermolecular energy and the ligand’s internal energy. Moreover, inhibition constant was calculated using the expression Ki=exp((ΔG*1000)/(Rcal*TK), where ΔG is docking energy, Rcal is 1.98719 and TK is 298.15 [35]. The values of binding energies, docking energies and inhibition constant for the selected compounds docked against Aurora kinases are listed (Table 1).
| Aurora kinase: | A | B | C | A | B | C | A | B | C | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | Binding energy (kcal/mol) | Docking energy (kcal/mol) | Ki (µM) | |||||||||
| a) | -6.18 | -7.1 | -6.06 | -7.43 | -7.99 | -6.96 | 16.08 | 6.28 | 36.04 | |||
| b) | -6.14 | -7.27 | -6.03 | -6.85 | -7.87 | -6.65 | 26.95 | 4.67 | 37.9 | |||
| c) | -6.91 | -7.74 | -7.63 | -6.91 | -7.74 | -7.63 | 8.62 | 2.1 | 2.55 | |||
| d) | -5.79 | -6.79 | -6.37 | -6.23 | -7.23 | -6.8 | 57.0 | 10.59 | 21.57 | |||
Table 1: Binding energies, docking energies and inhibition constant values for (a) 2-(thiophen-2-yl) quinazoline, (b) N-[(E)-phenylmethylidene]-1H-imidazole-1-carboxamide, (c) 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino} benzoic.
Altogether, binding energies of all four compounds ranged from -7.74 to -6.03 kcal/mol, while docking energies ranged from -7.99 to -6.23 kcal/mol. Figures 3a-3c shows docked model of 2-(thiophen-2- yl) quinazoline into the active sites of Aurora kinases. Quinazoline-Nring atom shows H-bonding with Ala213 of Aurora kinase A (-N...HN, distance; 1.9 Å). In addition, it exhibited hydrophobic interactions with Lys162, Glu211, Ala213 and Val147 (Figure 3a). Likewise, quinazoline -S- atom showed hydrophobic interactions with Leu83, and Val91. It also formulated pi-pi interactions with Tyr156 residue (Figure 3b). For Aurora kinase C, quinazoline-S-ringed atom showed hydrophobic interactions that were found with Leu49 and Ala70 residues (Figure 3c).
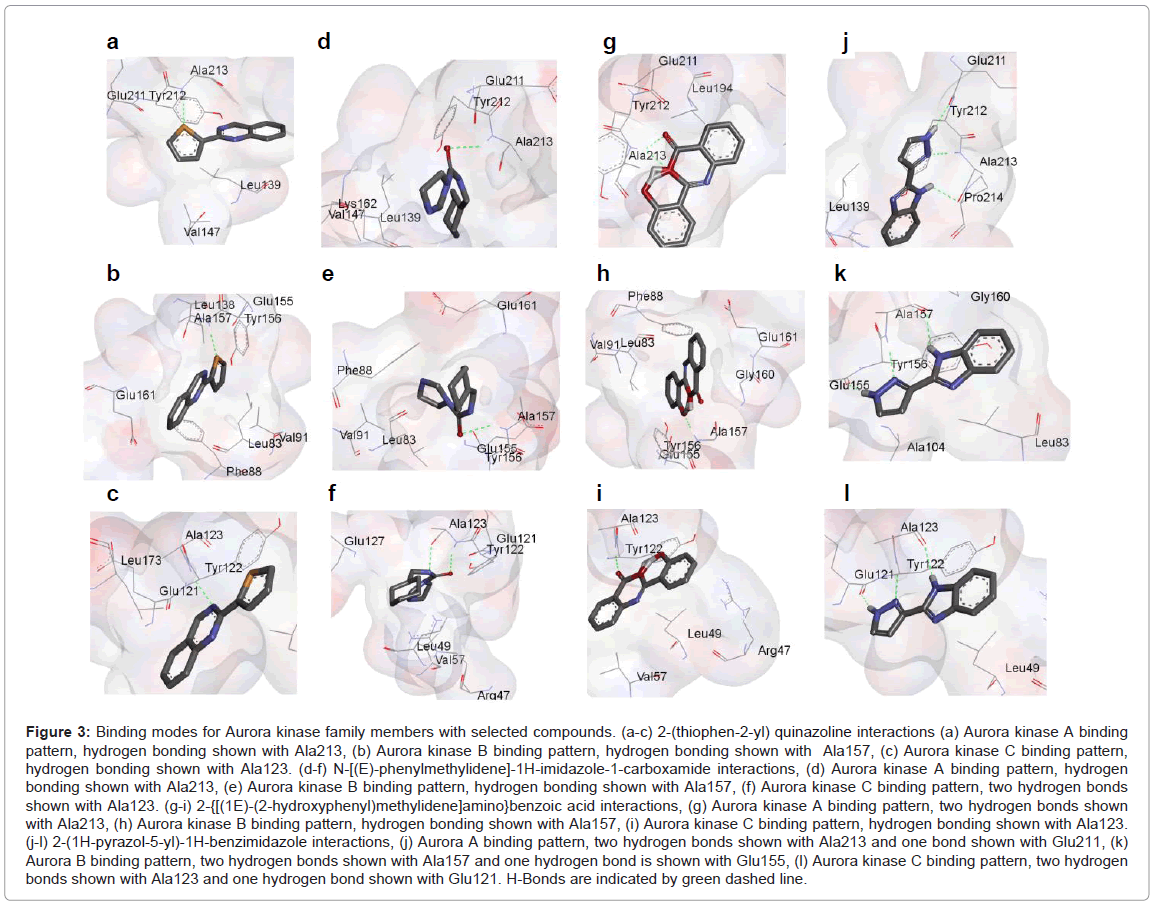
Figure 3: Binding modes for Aurora kinase family members with selected compounds. (a-c) 2-(thiophen-2-yl) quinazoline interactions (a) Aurora kinase A binding pattern, hydrogen bonding shown with Ala213, (b) Aurora kinase B binding pattern, hydrogen bonding shown with Ala157, (c) Aurora kinase C binding pattern, hydrogen bonding shown with Ala123. (d-f) N-[(E)-phenylmethylidene]-1H-imidazole-1-carboxamide interactions, (d) Aurora kinase A binding pattern, hydrogen bonding shown with Ala213, (e) Aurora kinase B binding pattern, hydrogen bonding shown with Ala157, (f) Aurora kinase C binding pattern, two hydrogen bonds shown with Ala123. (g-i) 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid interactions, (g) Aurora kinase A binding pattern, two hydrogen bonds shown with Ala213, (h) Aurora kinase B binding pattern, hydrogen bonding shown with Ala157, (i) Aurora kinase C binding pattern, hydrogen bonding shown with Ala123. (j-l) 2-(1H-pyrazol-5-yl)-1H-benzimidazole interactions, (j) Aurora A binding pattern, two hydrogen bonds shown with Ala213 and one bond shown with Glu211, (k) Aurora B binding pattern, two hydrogen bonds shown with Ala157 and one hydrogen bond is shown with Glu155, (l) Aurora kinase C binding pattern, two hydrogen bonds shown with Ala123 and one hydrogen bond shown with Glu121. H-Bonds are indicated by green dashed line.
Docked models for N-[(E)-phenylmethylidene]-1H-imidazole- 1-carboxamide with the Aurora kinases specific ATP binding site are shown (Figures 4d-4f). N-[(E)-phenyl methylidene]-1H-imidazole-1- carboxamide formulated H-bond with Ala213 of Aurora kinase A by its -O- atom (-O...HN, distance; 2.4 Å). In addition, its hydrophobic interactions were examined with Leu139, Val147, Lys162 and Leu194 residues (Figure 3d). The -O- atom of N-[(E)-phenylmethylidene]- 1H-imidazole-1-carboxamide exhibited H-bond with Ala157 of Aurora kinase B (-O...HN, distance; 2.4 Å). Furthermore, it showed hydrophobic interactions with Leu83, Val91, Glu155, Glu161 (Figure 3e). For Aurora kinase C, this compound represented two hydrogen bonds with Ala123 with -O- and -N- atoms (-O...HN, distance; 2.1 Å and N...HN, distance; 3.1 Å), whereas hydrophobic interactions were found with Leu49, Val57, Tyr122, Glu121 and Leu173 (Figure 3f).
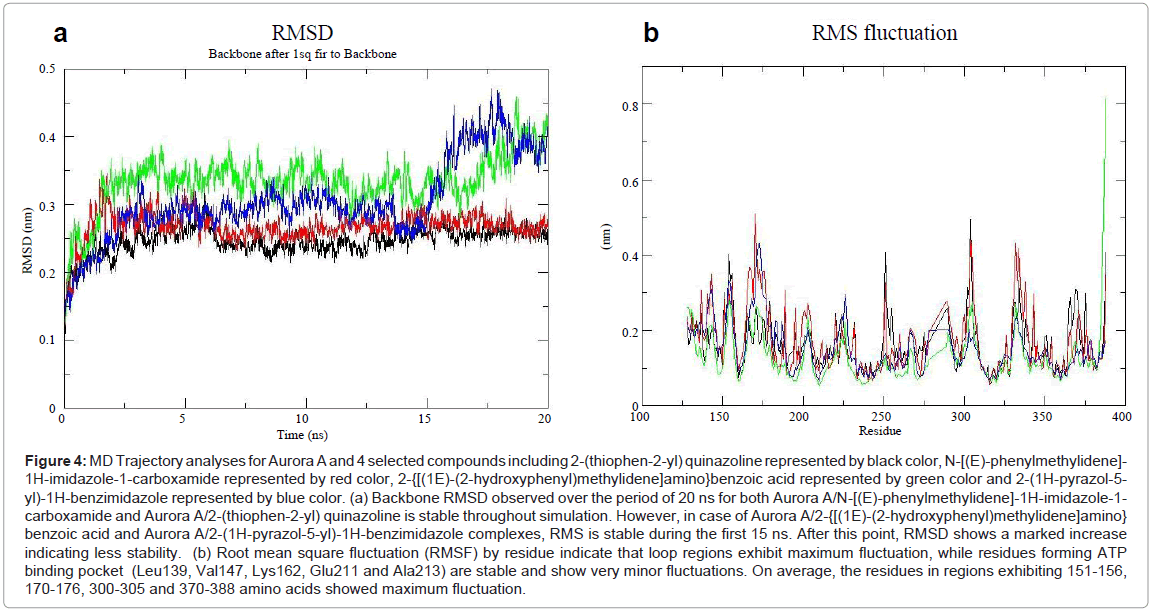
Figure 4: MD Trajectory analyses for Aurora A and 4 selected compounds including 2-(thiophen-2-yl) quinazoline represented by black color, N-[(E)-phenylmethylidene]- 1H-imidazole-1-carboxamide represented by red color, 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid represented by green color and 2-(1H-pyrazol-5- yl)-1H-benzimidazole represented by blue color. (a) Backbone RMSD observed over the period of 20 ns for both Aurora A/N-[(E)-phenylmethylidene]-1H-imidazole-1- carboxamide and Aurora A/2-(thiophen-2-yl) quinazoline is stable throughout simulation. However, in case of Aurora A/2-{[(1E)-(2-hydroxyphenyl)methylidene]amino} benzoic acid and Aurora A/2-(1H-pyrazol-5-yl)-1H-benzimidazole complexes, RMS is stable during the first 15 ns. After this point, RMSD shows a marked increase indicating less stability. (b) Root mean square fluctuation (RMSF) by residue indicate that loop regions exhibit maximum fluctuation, while residues forming ATP binding pocket (Leu139, Val147, Lys162, Glu211 and Ala213) are stable and show very minor fluctuations. On average, the residues in regions exhibiting 151-156, 170-176, 300-305 and 370-388 amino acids showed maximum fluctuation.
Figures 3g-3i shows complexes of 2-{[(1E)-(2-hydroxyphenyl) methylidene]amino}benzoic acid in Aurora kinases binding sites. 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid contained two H-bonds with Ala213 with its two -O- atoms(-O...HN, distance; 2 Å and -O...HN, distance; 2.5 Å), whereas hydrophobic interactions were found with Ala160, Leu210, Glu211, Pro214, Gly216, Lys162 and Leu263 residues (Figure 3g). The pi-pi interaction was examined with Tyr212. For Aurora kinase B, 2-{[(1E)-(2-hydroxyphenyl)methylidene] amino}benzoic acid showed hydrogen bonding with Ala157 through its -O- atom (-O...HN, distance; 2 Å) while hydrophobic interactions were with Leu83, Val91, Gly160, Glu155, Glu161 residues (Figure 3h). Two pi-pi interactions were also detected with Phe88 and Tyr156 residues. For Aurora kinase C, this compound showed hydrogen bonding with Ala123 via its -O- atom (-O...HN, distance; 1.8 Å) while hydrophobic interactions were mapped with Val57, Leu49 Tyr122, Pro124, and Gly126 residues. In contrast, the pi-pi interaction was found with Arg47 (Figure 3i).
Docked complexes for 2-(1H-pyrazol-5-yl)-1H-benzimidazole with Aurora kinases binding sites are indicated in (Figure 3j) -l. 2-(1H-pyrazol-5-yl)-1H-benzimidazole had two hydrogen bonds with Ala213 via its -N- and -NH- atoms (-N...HN, distance; 2.1 Å and -NH...O, distance; 1.8 Å) and one hydrogen bond through Glu211 with its -NH- atom (-NH...O, distance; 2 Å). On the other hand, it showed hydrophobic interactions with Arg137, Leu139, Ala160 and Leu194 residues (Figure 3j). For Aurora kinase B, this compound formulated two bonds with Ala157 by -N- and -NH- atoms (-N...HN, distance; 2.1 Å -NH...O and distance; 2 Å), and one hydrogen bond with Glu155 via its -NH- atom (-NH...O, distance;1.9 Å). Moreover, it showed hydrophobic interactions with Leu83, Ala104, Leu138, Leu207 and Gly160 (Figure 3k). The pi-pi interaction was detected with Tyr156. For Aurora kinase C, this compound formulated two hydrogen bonds with Ala123 via its -N- and -NH- atoms (-N...HN, distance; 2 Å and -NH...O, distance; 1.9 Å) and one hydrogen bond with Glu121 through its -NH- atoms (-NH...O, distance; 2.1 Å), whereas hydrophobic interactions were detected with Glu121, Tyr122 and Gly126 residues. The pi-pi interactions were identified with Arg47 (Figure 3).
MD simulations
Experimentally defined biological activities can be efficiently validated by MD simulations in order to search interaction network. Structure based VS provided the binding and orientation of novel inhibitors including 2-(thiophen-2-yl) quinazoline, N-[(E)-phenylmethylidene]- 1H-imidazole-1-carboxamide, 2-{[(1E)-(2-hydroxyphenyl) methylidene]amino} benzoic acid and 2-(1H-pyrazol-5-yl)- 1H-benzimidazole at ATP binding site of Aurora kinases. The predicted complexes were then studied in context of dynamics by analyzing their time dependent behavior in the form of MD trajectories. Each of the four molecular models was used to run 20 ns simulation at constant temperature and pressure under periodic boundary conditions. The structural intermediates which led the protein to change its active site conformations were investigated against their respective inhibitor.
In order to monitor the conformational changes and stability of secondary structure elements, we assessed the backbone root mean square deviation (RMSD) scores derived from the simulated complexes of Aurora A and each of the compound including 2-(thiophen-2-yl) quinazoline, N-[(E)-phenylmethylidene]-1H-imidazole-1-carboxamide, 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid and 2-(1H-pyrazol-5-yl)-1H-benzimidazole (Figure 5a). Backbone RMSD score observed over the period of 20 ns for both Aurora A/N-[(E)- phenylmethylidene]-1H-imidazole-1-carboxamide and Aurora A/2- (thiophen-2-yl) quinazoline was stable throughout simulation. However, in case of Aurora A/2-{[(1E)-(2-hydroxyphenyl)methylidene]amino} benzoic acid and Aurora A/2-(1H-pyrazol-5-yl)-1H-benzimidazole complexes, RMS was stable during the first 15 ns. After this point, RMSD showed a marked increase indicating less stability (Figure 4a). In contrast, Root Mean Square Fluctuation (RMSF) by residue indicated maximum fluctuations at the loop regions, while residues lying at ATP binding pocket (Leu139, Val147, Lys162, Glu211 and Ala213) were quite stable and exhibited minor fluctuations. Moreover, α-helices and β-strands were observed much stable (Figure 4b).
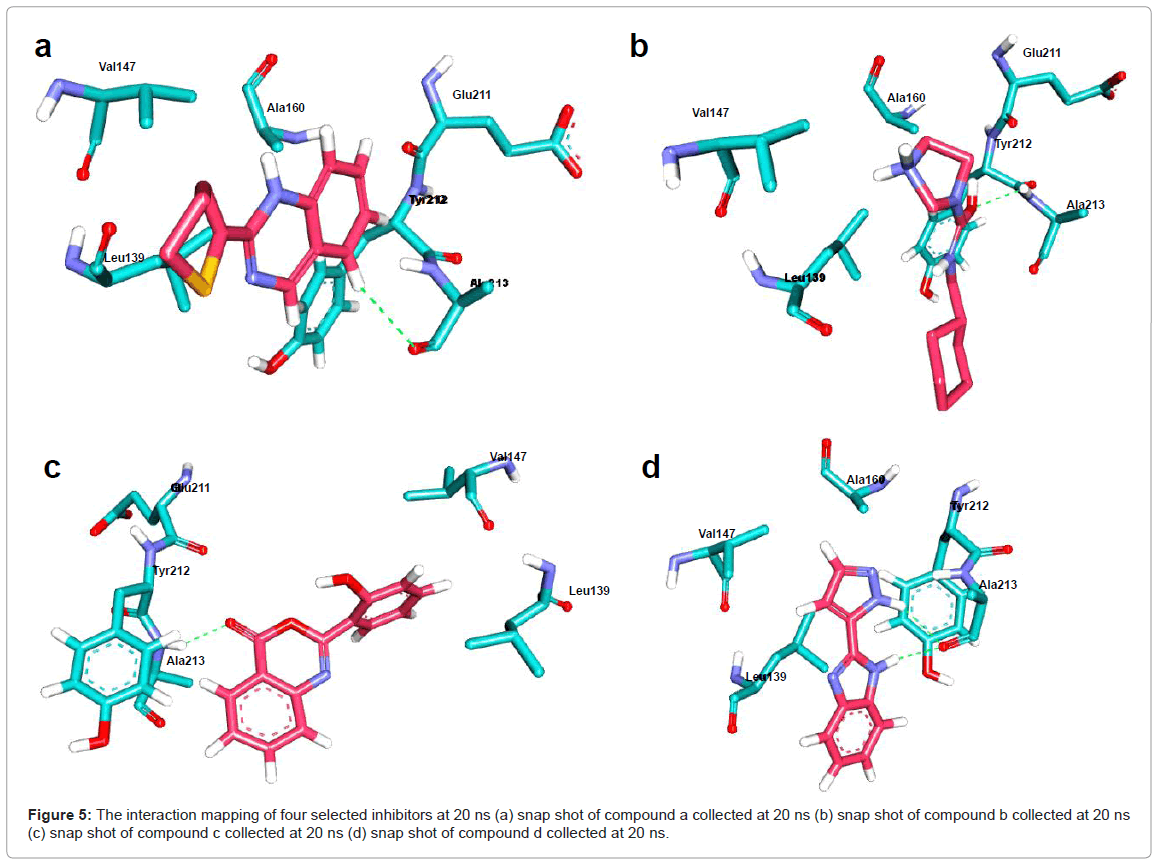
Figure 5: The interaction mapping of four selected inhibitors at 20 ns (a) snap shot of compound a collected at 20 ns (b) snap shot of compound b collected at 20 ns (c) snap shot of compound c collected at 20 ns (d) snap shot of compound d collected at 20 ns.
The visual inspection of trajectories was done using VMD, which clearly showed the binding of all four inhibitors at the ATP binding pocket (Figure 5). The number of hydrogen bond interactions between ATP binding pocket and inhibitors was also measured throughout the time of simulations. It was observed that Ala213 is very crucial amino acid that formed necessary H-bond interactions with selected compounds (Figures 5a-5d). Intriguingly, interactions of Leu139, Val147, Lys162, Glu211, Ala160 and Tyr212 residues lying at ATP binding site were quite conserved. Given that the selected compounds remained intact at 20 ns simulation time with Aurora A specific ATP binding pocket, these structures selected from MD trajectories were used in the generation of structure-based pharmacophore models.
Evaluation of pharmacophore mode
Ten pharmacophore models were generated by ligand-based pharmacophore modeling strategy using four ligand dataset 2-(thiophen-2-yl) quinazoline, N-[(E)-phenylmethylidene] -1Himidazole-1-carboxamide, 2-{[(1E)-(2-hydroxyphenyl)methylidene] amino}benzoic acid and 2-(1H-pyrazol-5-yl) -1H-benzimidazole). The scores of these models along with their corresponding features are listed (Table 2). Based on the essential amino acids lying at the ATP binding site, clusters of features were generated. The pharmacophoric features in all cases were made of 1 HBA, 1 HBD, 2 AR and 1 HD features complementing the same set of amino acid residues (Table 2). Among all pharmacophore models, model 1 was chosen on the basis of selective features and high score value, and was subsequently used as a reference for screening Princeton and Uorsy kinase databases consisting of 50,000 and 15,000 structurally diversified molecules in order to retrieve novel scaffold of Aurora kinase inhibitors. The geometrical relationship among pharmacophore features is indicated (Figure 6a). Though all pharmacophore models were generated using the same features, the inter-feature distance constraints of these models were different (Figure 6b). (Figures 6c-6f) shows alignment of four ligands with pharmacophore features.
| Models | Score | HBA | HBD | AR | HD |
|---|---|---|---|---|---|
| 1 | 0.6503 | + | + | ++ | + |
| 2 | 0.6200 | + | _ | ++ | + |
| 3 | 0.6180 | + | _ | ++ | + |
| 4 | 0.6169 | ++ | _ | + | + |
| 5 | 0.6168 | ++ | _ | + | + |
| 6 | 0.6166 | + | + | + | + |
| 7 | 0.6166 | + | + | + | + |
| 8 | 0.6157 | ++ | _ | + | _ |
| 9 | 0.6121 | ++ | _ | + | _ |
| 10 | 0.6118 | ++ | + | _ | + |
Table 2: Scores of ten pharmacophore models along with their features. HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; AR, aromatic ring; HD, hydrophobic.
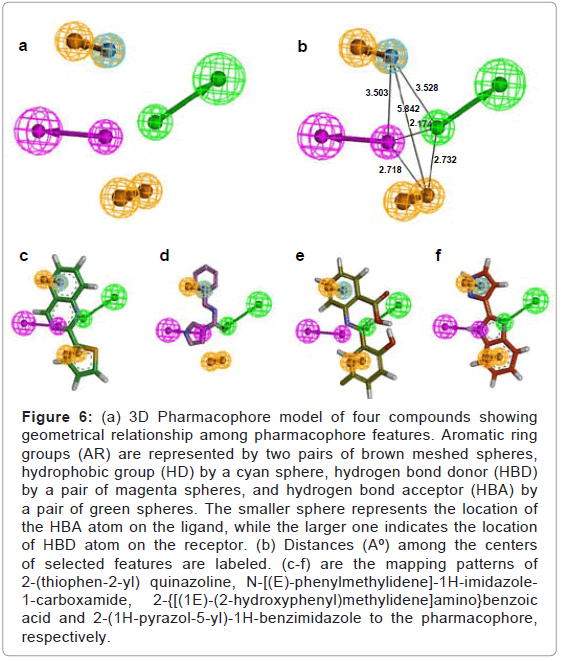
Figure 6: (a) 3D Pharmacophore model of four compounds showing geometrical relationship among pharmacophore features. Aromatic ring groups (AR) are represented by two pairs of brown meshed spheres, hydrophobic group (HD) by a cyan sphere, hydrogen bond donor (HBD) by a pair of magenta spheres, and hydrogen bond acceptor (HBA) by a pair of green spheres. The smaller sphere represents the location of the HBA atom on the ligand, while the larger one indicates the location of HBD atom on the receptor. (b) Distances (Aº) among the centers of selected features are labeled. (c-f) are the mapping patterns of 2-(thiophen-2-yl) quinazoline, N-[(E)-phenylmethylidene]-1H-imidazole- 1-carboxamide, 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid and 2-(1H-pyrazol-5-yl)-1H-benzimidazole to the pharmacophore, respectively.
Virtual screening of princeton database
Selected pharmacophore model was then used as 3D query to screen the compound libraries. Princeton library comprising of 50,000 compounds was screened and 13,982 compounds were identified that shared pharmacophore-like features. The extracted compounds were refined using various filters like Lipinski’s rule. 180 compounds were extracted on the basis of exact pharmacophore features including one H-acceptor, one H-donor, two aromatic rings and one hydrophobic region. These 180 compounds were subsequently used for docking analysis. Next, receptor and ligand complementarities were checked and 160 compounds were filtered on the basis of these criteria. Compounds having positive binding energies were eliminated leaving 130 compounds for further study. Interactions were mapped for these compounds with Aurora family members and 30 compounds were short listed based on frequent interaction with ATP binding site (Table 3). 2D structures of selected hits are indicated (Figure 7), while binding energy, docking energy and inhibition constant values for these hits are shown in table 3.
| Aurora kinase: | A | B | C | A | B | C | A | B | C |
|---|---|---|---|---|---|---|---|---|---|
| Compounds | Binding Energy (kcal/mol) | Docking Energy (kcal/mol) | Inhibition Constant (µM) | ||||||
| 1 | -8.42 | -8.13 | -7.48 | -9.87 | -9.52 | -8.75 | 0.668 | 1.11 | 3.28 |
| 2 | -8.55 | -9.33 | -8.82 | -9.99 | -10.35 | -9.01 | 0.538 | 0.143 | 0.342 |
| 3 | -8.21 | -8.85 | -7.55 | -9.35 | -10.34 | -9.11 | 0.958 | 0.324 | 2.92 |
| 4 | -7.35 | -8.8 | -7.77 | -9.83 | -11.09 | -10.54 | 4.13 | 0.356 | 2.03 |
| 5 | -8.96 | -8.47 | -8.48 | -10.5 | -9.77 | -9.46 | 0.269 | 0.620 | 0.612 |
| 6 | -8.18 | -10.36 | -9.15 | -10.18 | -12.38 | -11.17 | 1.01 | 0.25 | 0.196 |
| 7 | -6.27 | -7.37 | -6.31 | -8.92 | -9.99 | -9.12 | 29.8 | 3.95 | 11.28 |
| 8 | -7.88 | -8.78 | -9.59 | -9.54 | -10.37 | -10.02 | 1.67 | 0.365 | 0.93 |
| 9 | -7.08 | -7.33 | -6.66 | -9.25 | -9.84 | -9.17 | 6.5 | 4.23 | 13.2 |
| 10 | -6.41 | -7.17 | -6.16 | -7.57 | -9.07 | -7.78 | 20.17 | 5.53 | 0.30 |
| 11 | -7.66 | -7.95 | -8.96 | -8.39 | -8.67 | -9.7 | 2.42 | 1.5 | 0.270 |
| 12 | -8.06 | -8.76 | -8.93 | -8.29 | -9.33 | -9.44 | 1.23 | 0.381 | 0.286 |
| 13 | -9.29 | -10.29 | -8.88 | -10.17 | -11.79 | -10.34 | 0.154 | 0.28 | 0.307 |
| 14 | -8.45 | -9.04 | -9.3 | -9.91 | -10.59 | -10.81 | 0.639 | 0.236 | 0.152 |
| 15 | -8.74 | -9.73 | -10.01 | -8.97 | -9.55 | -9.26 | 0.392 | 0.73 | 0.45 |
| 16 | -8.01 | -10.11 | -8.8 | -10.22 | -12.41 | -11.15 | 1.34 | 0.38 | 0.355 |
| 17 | -7.71 | -8.78 | -7.56 | -9.04 | -9.96 | -8.68 | 2.22 | 0.366 | 2.86 |
| 18 | -8.3 | -10.2 | -9.08 | -10.59 | -12.62 | -11.15 | 0.827 | 0.33 | 0.222 |
| 19 | -9.2 | -9.96 | -10.18 | -10.22 | -10.96 | -10.9 | 0.181 | 0.50 | 0.34 |
| 20 | -9.3 | -10.96 | -9.87 | -10.2 | -12.4 | -11.13 | 0.15 | 0.9 | 0.58 |
| 21 | -8.21 | -9.86 | -8.55 | -10.33 | -12.14 | -10.81 | 0.957 | 0.59 | 0.536 |
| 22 | -9.1 | -10.66 | -9.09 | -11.17 | -12.63 | -10.05 | 0.213 | 0.15 | 0.216 |
| 23 | -8.09 | -9.12 | -8.56 | -9.96 | -10.84 | -10.5 | 1.17 | 0.205 | 0.534 |
| 24 | -9.04 | -10.39 | -9.5 | -9.94 | -11.5 | -9.89 | 0.238 | 0.24 | 0.109 |
| 25 | -7.59 | -9.2 | -9.07 | -10.13 | -11.36 | -11.49 | 2.73 | 0.181 | 0.223 |
| 26 | -7.23 | -8.64 | -7.63 | -8.96 | -10.37 | -9.29 | 4.99 | 0.466 | 2.57 |
| 27 | -8.92 | -9.6 | -9.46 | -9.11 | -9.85 | -9.7 | 0.288 | 0.92 | 0.117 |
| 28 | -8.57 | -9.49 | -9.35 | -8.67 | -9.48 | -9.07 | 0.521 | 0.110 | 0.139 |
| 29 | -8.76 | -9.68 | -10.02 | -9.77 | -10.58 | -10.48 | 0.381 | 0.80 | 0.45 |
| 30 | -15.3 | -16.3 | -15.02 | -16.1 | -16.94 | -15.83 | 6X10-6 | 1.13 | 0.985 |
Table 3: Binding energies, docking energies and inhibition constant values for compounds bound to Aurora kinase A, B and C isolated from Princeton library.
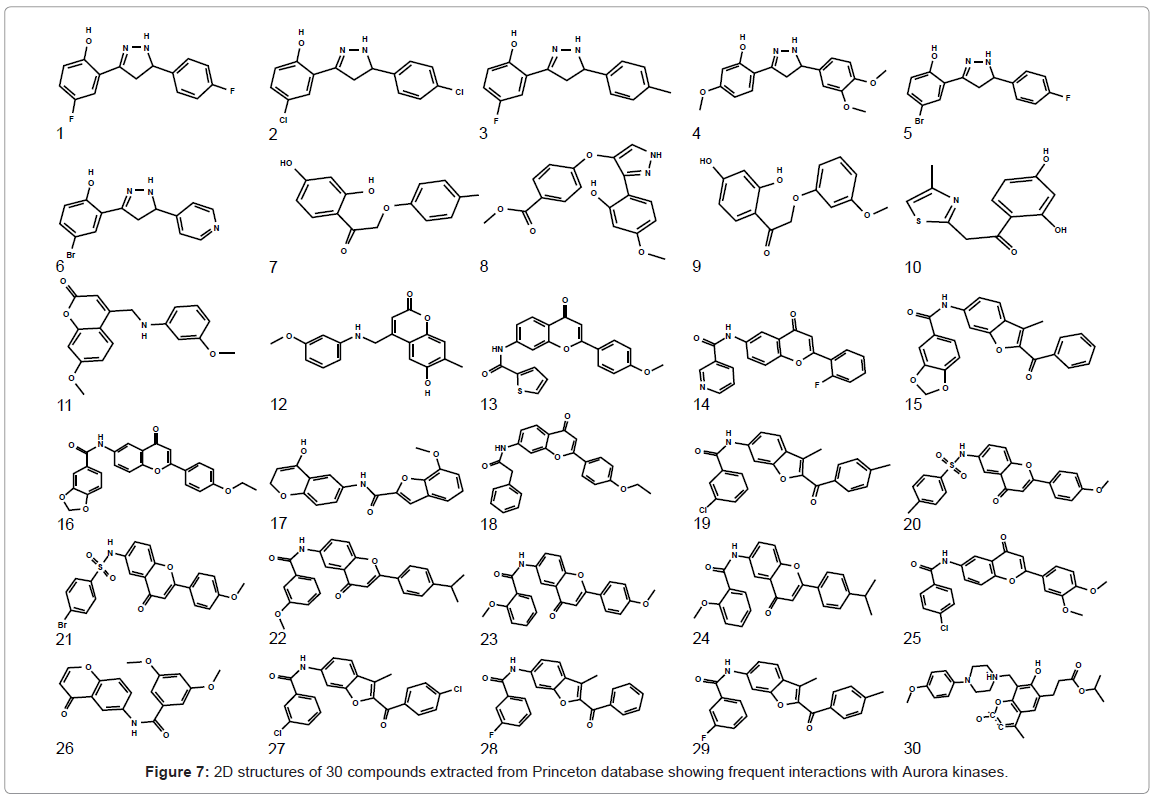
Figure 7: 2D structures of 30 compounds extracted from Princeton database showing frequent interactions with Aurora kinases.
Molecular docking studies for princeton screened hits
To reduce the false positives, retrieved hits were evaluated using molecular docking analyses. The docked compounds were refined based on binding mode, docking score, and molecular interactions with the ATP binding site. For Aurora A binding compounds, binding energy values ranged from -6.41 to -15.3 kcal/mol, while the respective docking energies were in the range of -7.57 to -16.1 kcal/mol. Intermolecular energy values ranged from -7.6 to -16.19 kcal/mol, while unbound energy values ranged from 0 to -1.05 kcal/mol. Similarly, Aurora B bound compounds exhibited binding energies ranging from -7.17 to - 6.3 kcal/mol. The corresponding docking energies ranged from -8.67 to -16.94 kcal/mol. Furthermore, intermolecular energies were from -8.37 to -17.9 kcal/mol. For Aurora C, binding energies ranged from -6.16 to -15.02 kcal/mol, whereas docking energies ranged from -7.78 to -15.83 kcal/mol, while intermolecular energies ranged from -7.35 to -15.91 kcal/mol (Table 3). Out of these hits, 1-6 belong to phenol family, 7-8 are from benzoate family, 9-10 are from ethanone family, 11-12 are from chromen-2-one family, 13-19 are from carboxamide family, 20-22 are from sulfonamide family, 23-29 are from benzamide family, while compound 30 belong to propanoate family. Among these, compound-30 was detected to have lowest binding energy, docking energy and inhibition constant values; this compound was used for in detail pharmacophore features and interaction with Aurora proteins. Mapping of this hit compound with pharmacophore features along with detailed interaction analyses with Aurora A, B and C are indicated (Figure 8). In Aurora A, compound-30 showed hydrogen bonding with Ala213 through its -O- atom (-O...HN, distance; 2Å), while it formed hydrophobic interactions via Val147, Lys162 and Glu211 residues. In addition, 3 pi-pi interactions were detected with Try212, Arg137, Leu139 residues. In contrast, for Aurora B, this hit showed hydrogen bonding with Ala157 (-O...HN, distance; 1.8Å) via its -Oatom, hydrophobic interactions with Arg81, Phe88, Val91, Lys106 and Glu155 residues. The pi-pi interaction was found with Try156. In case of Aurora C, compound-30 bonding was detected with Ala123 by its -O- atom (-O...HN, distance; 2Å), while it formulated hydrophobic interactions with Lys72, Glu121, Gly12 and Leu173 residues. The pi-pi interactions were identified with Arg47 and Leu173 (Figure 8).
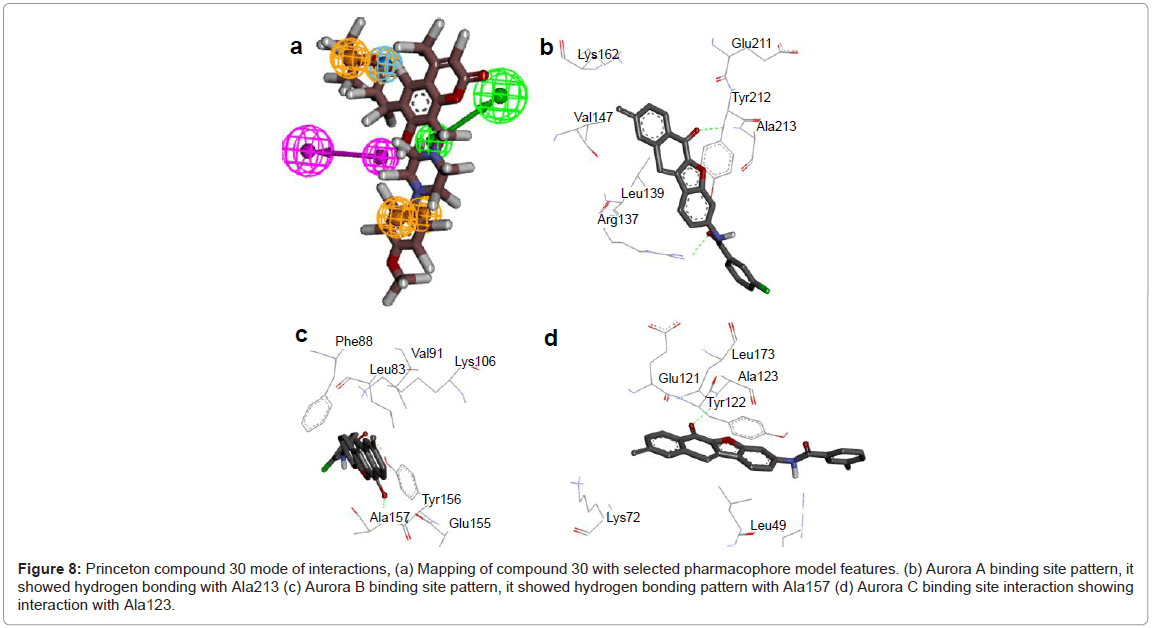
Figure 8: Princeton compound 30 mode of interactions, (a) Mapping of compound 30 with selected pharmacophore model features. (b) Aurora A binding site pattern, it showed hydrogen bonding with Ala213 (c) Aurora B binding site pattern, it showed hydrogen bonding pattern with Ala157 (d) Aurora C binding site interaction showing interaction with Ala123.
Virtual screening of uorsy database
Next, a library of 15,000 compounds derived from Uorsy database was screened using the generated pharmacophore hypothesis and 10,330 compounds were short listed, based on the common pharmacophore-like features. Subsequently, 380 compounds having the exact pharmacophore-like features were selected for docking studies and on the basis of receptor and ligand complementarities, 250 compounds were filtered. Finally, 50 compounds were tested for binding studies with Aurora kinases and 12 compounds were refined on the basis of binding energy values. 2D structures of these compounds were shown (Figure 9), while binding energies, docking energies and inhibition constant were shown (Table 4).
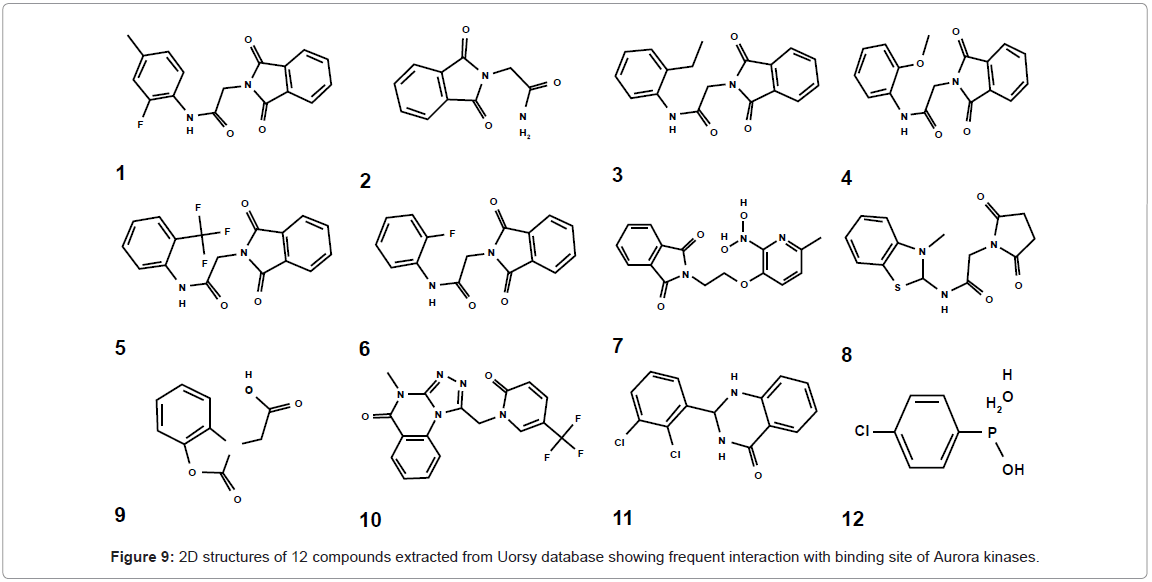
Figure 9: 2D structures of 12 compounds extracted from Uorsy database showing frequent interaction with binding site of Aurora kinases.
| Aurora kinase: | A | B | C | A | B | C | A | B | C |
|---|---|---|---|---|---|---|---|---|---|
| Compounds | Binding energy (kcal/mol) | Docking energy (kcal/mol) | Inhibition constant (µM) | ||||||
| 1 | -8.87 | -9.9 | -9.05 | -9.46 | -10.49 | -9.65 | 0.317 | 0.055 | 0.232 |
| 2 | -7.1 | -7.21 | -6.46 | -8.95 | -9.07 | -8.3 | 6.23 | 5.21 | 18.42 |
| 3 | -9.76 | -11.45 | -10.44 | -10.06 | -11.75 | -10.74 | 0.07 | 0.004 | 0.022 |
| 4 | -10.37 | -11.54 | -10.56 | -10.67 | -11.84 | -10.85 | 0.025 | 0.004 | 0.018 |
| 5 | -9.3 | -10.89 | -9.83 | -9.6 | -11.19 | -10.13 | 0. 152 | 0.01 | 0.062 |
| 6 | -9.24 | -10.45 | -9.58 | -9.54 | -10.74 | -9.88 | 0.167 | 0.022 | 0.094 |
| 7 | -9.5 | -10.79 | -9.94 | -9.8 | -11.09 | -10.24 | 0.109 | 0.012 | 0.051 |
| 8 | -10.02 | -10.55 | -9.5 | -10.02 | -10.55 | -9.5 | 0.045 | 0.019 | 0.110 |
| 9 | -6.81 | -6.61 | -6.03 | -7.73 | -7.52 | -6.95 | 10.21 | 14.35 | 38.08 |
| 10 | -8.04 | -8.55 | -8 | -8.93 | -9.45 | -8.89 | 1.27 | 0.536 | 1.37 |
| 11 | -7.71 | -7.33 | -6.59 | -7.71 | -7.33 | -6.59 | 2.25 | 4.27 | 14.87 |
| 12 | -8.77 | -9.84 | -9.2 | -8.77 | -9.84 | -9.2 | 0.374 | 0.061 | 0.181 |
Table 4: Binding Energies, docking energies and inhibition constant values for Aurora A, B and C bound compounds isolated from Uorsy database.
Binding mode studies for uorsy screened compounds
Binding, docking and intermolecular energy values for Uorsy screened hits with Aurora A, B and C are indicated. Among these hits, 1-8 belong to acetamide family, number 9 belongs to acetic acid family, 9-11 belong to quinazoline family, while compound 12 belongs to phosphane family. All these selected compounds exhibited frequent interactions with Aurora kinase specific ATP binding site. Compound 4 belonging to acetamide family showed lowest binding and docking energies among all selected compounds. Mapping of this compound with developed pharmacophore features and interaction with Aurora proteins were analyzed in detail and indicated (Figure 10). For Aurora kinase A, compound 4 was having H-bond with Ala213 by its -O- atom (-O...HN, distance; 1.9Å), hydrophobic interactions with Lys162, Glu211, Val147and Leu139 residues. For this compound, Aurora kinase B showed two H-bonds with -O- atom of Ala157 (-O... HN, distance; 2.7Å) and Glu161 (-O...HN, distance; 3.7Å), while one H-Bond was via its -H- atom with Leu83 (-H...O, distance; 2.7Å). In contrast, hydrophobic interactions were with Phe88 and Tyr156. For Aurora kinase C, compound 4 was having H-Bond with Ala123 with -O- atom (-O...HN, distance; 2.1Å). The hydrophobic interactions were observed with Leu49, Val57, Lys72 and Arg47 residues (Figure 10).
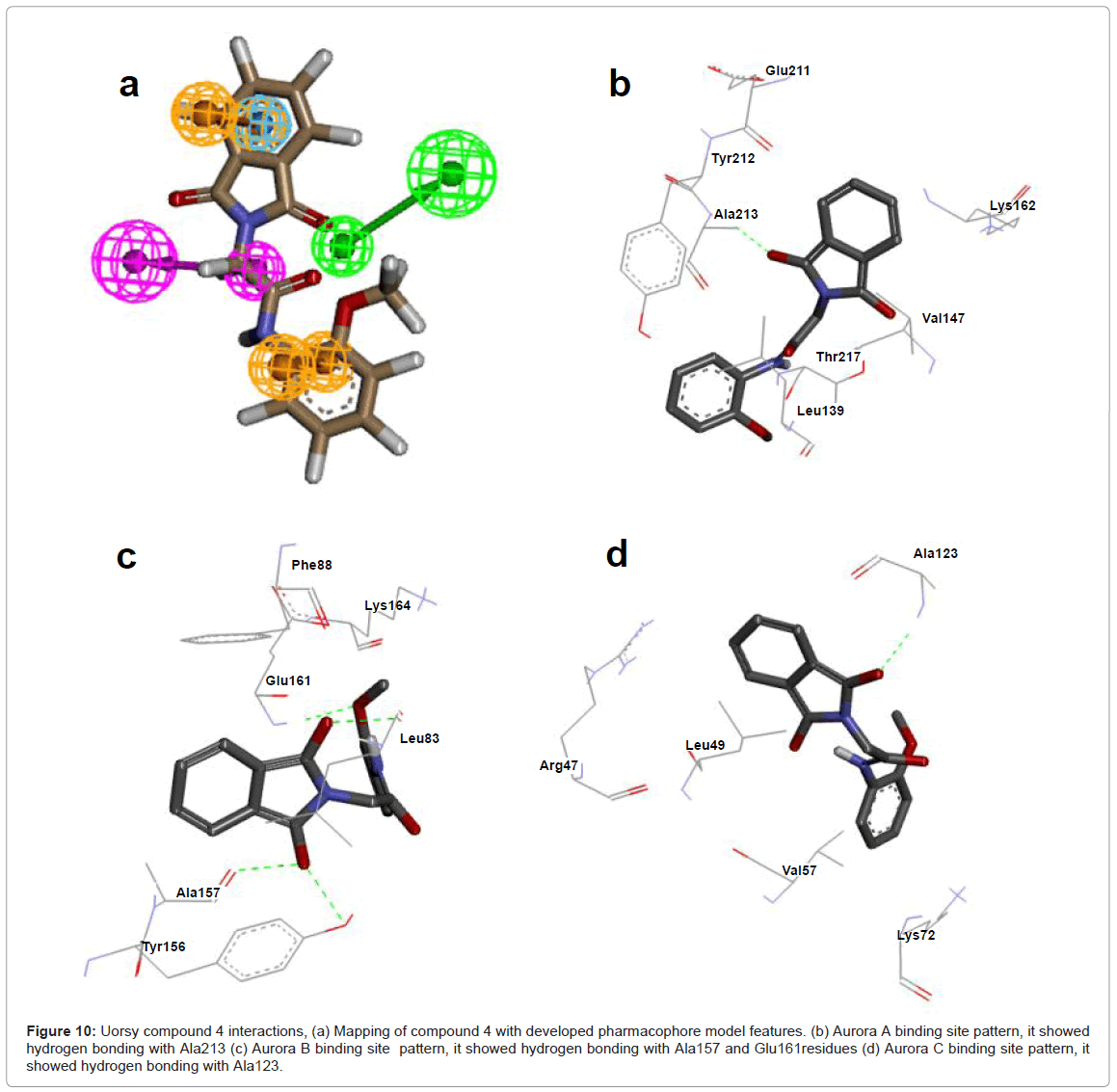
Figure 10: Uorsy compound 4 interactions, (a) Mapping of compound 4 with developed pharmacophore model features. (b) Aurora A binding site pattern, it showed hydrogen bonding with Ala213 (c) Aurora B binding site pattern, it showed hydrogen bonding with Ala157 and Glu161residues (d) Aurora C binding site pattern, it showed hydrogen bonding with Ala123.
Aurora kinases are very important regulators, as they are implicated in multiple tumor types and diverse cell cycle events [1- 8]. In this study, it was initially characterized four representative Aurora specific inhibitors, including I: 2-(thiophen-2-yl) quinazoline, II: N-[(E)-phenylmethylidene]-1H-imidazole-1-carboxamide; III: 2-{[(1E)-(2-hydroxyphenyl)methylidene]amino}benzoic acid and a known compound, IV: 2-(1H-pyrazol-5-yl)-1H-benzimidazole [47] on the basis of their comparative binding to ATP binding site of Aurora proteins. Following the validation of their binding, these test set compounds were used for pharmacophore model generation by ligand- based strategy. Generally, derived pharmacophore models from a set of ligands in the absence of a macromolecule structure, are based on the principle that common structures containing small molecules exhibit similar biological activity. Thus, this approach searches a common feature pattern that is shared in an active ligand-set [44].
Previous studies reported various inhibitors based on the established quinazoline core, where the selectivity of inhibitors has been increased by introduction of a large lipophilic group and substitutions by methylene spacer [48]. Similarly, substitution by aminopyrazole in quinazoline-based inhibitors led to profound increase in its activity [49], as aminopyrazole mimics the adenosine substrate of ATP [50]. However, limitation in this context is the possible involvement of quinazoline-pyrazole series compounds in potential inhibition of PDGFRα, PDGFRβ, CST-1R and c-KIT kinases, along with Aurora B [49]. The compound I, isolated in our study contains imidazole moiety, which is involved in the stability of coplanar state by existence of internal H-bond between the imidazole NH and the quinazoline N3. Our results show that quinazoline ring adopts a strongly preferred orientation during interaction with Aurora A, by making hydrogen bond (-N...HN, distance; 1.9 Å). Likewise, the corresponding sulfur atom located on the same side is adjusted to further stabilize this interaction.
Our model for compound II belongs to carboxamide moiety, which is a well known category of ATP-binding site targeting inhibitor for many enzymes [51]. Our docking analyses indicated a pronounced binding of compound II with variants of Aurora kinase, due to possible involvement of its -O- atom in H-bonding with Ala213 (Aurora A) and Ala157 (Aurora B) residues (-O...HN, distance; 2.4 Å). In case of Aurora Kinase C, another H-bond was observed between its -N- and HN- of Ala123 residues at a distance of 3.1 Å. Similar patterns of Ala residues in H-bonding were observed for models III and IV, by binding analyses of benzoic acid and benzimidazole derivatives and Aurora proteins. Notably, the involvement of Ala213, Ala157 and Ala123 residues (located at hinge region of Aurora kinase) in H-bonding is dominant at the ATP-binding site, which compete with catalytic binding of ATP. Both Ala213 and Ala157 have been reported to be critical residues, involved in stabilized H-bonding with multiple inhibitors [45,46,52].
In order to obtain energetically favorable conformation for pharmacophore generations, the models of Aurora kinases and set compounds were subjected to MD simulation. We observed a stable binding mode for these inhibitors by monitoring RMSD, RMSF and Rg values at ATP-binding region. The best pharmacophore model was chosen as 3D query to screen the Princeton and Uorsy databases and the retrieved compounds were filtered by applying filters like Lipinski’s rule of five and ADMET properties. Hits showing exact pharmacophore-like features were further short-listed and subjected to docking analyses against Aurora proteins. Hence, 42 hits identified in this study by pharmacophore features exhibited exactly matched docking conformations to the ATP-binding site. This knowledge will be useful to accurately predict the chemical features for interacting analogues and help in designing novel Aurora specific inhibitors with improved activity.
This research was supported by Higher Education Commission, Pakistan (Grants No. 20-1493/R&D/09). SB performed major in silico analysis and compiled figures. SF conducted MD simulations. MK assisted in drafting the manuscript and supported the data by cross-validation. HI performed virtual screening. SR conceived and supervised this study and drafted the manucript. All authors (Batool, Saba Ferdous, Mohammad A. Kamal, Hira Iftikhar and Sajid Rashid) read and approved the final manuscript.