Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2012) Volume 5, Issue 4
Protein structure determination is crucial to understand a protein’s function and to develop drugs against diseases. Nuclear Magnetic Resonance (NMR) spectroscopy is an experimental technique that allows one to study protein structure in solution. In NMR Structure-Based Assignment (SBA) problem, the aim is to assign experimentally observed peaks to the specific nuclei of the target molecule by using a template protein and it is an important computational challenge. NVR is an NMR SBA framework in which multiple types of NMR data are combined to compute the assignments. In this paper, we studied the effect of incorporating additional sources of data into NVR. We added two types of data, chemical shifts for atoms other than 15N and HN, or HADAMAC experiment. We utilized an amino acid typing software Craack, that takes the chemical shifts of C, N and H atoms and returns the possible amino acids along with their confidence scores. This approach resulted in improved assignment accuracies. HADAMAC experiment which helps to predict an amino acid class for each peak was also incorporated into NVR, with improved assignment accuracies.
Keywords: NMR; Structure-based assignments; Protein structure
NMR: Nuclear Magnetic Resonance; CS: Chemical Shift; RDC: Residual Dipolar Coupling; NOE: Nuclear Overhauser Effect; SBA: Structure-Based Assignment; NVR: Nuclear Vector Replacement; BIP: Binary Integer Programming; SVM: Support Vector Machine
Proteins are one of the major macromolecules that are present in all biological organisms. They serve as enzymes, used as storage molecules, needed for the immune system and have many other functions in the cell. Determining the functions of proteins is crucial to understand important biological processes and to develop drugs against diseases. The function of a protein depends on its 3-D structure. There are two main experimental methods to determine the protein structure. These are X-ray crystallography and Nuclear Magnetic Resonance (NMR) Spectroscopy. About 85% of the protein structures in the Protein Data Bank were determined using X-ray Crystallography, on the other hand approximately 15% were solved using NMR. NMR allows one to study protein structure in solution. In addition, not all proteins can be crystallized. Therefore, NMR spectroscopy is an important experimental technique for protein structure determination.
In NMR, several experiments are performed on the protein and the signals are recorded. After processing these signals, the experiments result in various NMR spectra. The initial stage is to pick the peaks in the NMR spectrum and this stage is largely automated. The second stage is to find the mapping between the peaks and the atoms. This is called the assignment problem and is an important computational challenge. An existing structure (the “template”) can be used to help assign a target protein. This is called Structure-Based Assignment (SBA). SBA is analogous to molecular replacement in X-ray Crystallography [1].
In NMR SBA, the data coming from NMR spectroscopy and the template protein are analyzed. The available programs use a scoring function that maps each (peak, amino acid) pair to a real number that corresponds to the likelihood of the corresponding assignment. Then various methods (such as Monte Carlo Simulation, memetic algorithm or integer programming) are employed to find the assignments corresponding to the optimum or near-optimum of this scoring function (see e.g. MONTE [2], MATCH [3], NVR-BIP [4]).
The accuracy of NVR-BIP is highly related to the quality of the scoring function. Therefore, improving the scoring function will improve the assignment accuracies. This can be achieved by incorporating additional experimental data into NVR. For instance, additional chemical shifts obtained from triple resonance experiments can be added to NVR’s data types. These chemical shifts could then be used with amino acid typing to help determine the type of the amino acids or reduce the possibilities, therefore act as a filter.
Amino acid typing refers to the determination of the amino acid type based on NMR data, such as chemical shifts can be used as a filter to help in NMR assignments. Craack [7] is an amino acid typing tool that combines multiple programs to help determine the amino acid type. Another approach is to utilize the HADAMAC [8] experiment which uses Hadamard encoded amino acid type editing scheme. In Hadamard encoded type editing, the twenty amino acids are grouped into seven classes.
The main contributions of this work are as follows:
(1) We used amino acid typing software Craack to predict the amino acid groups that each NMR peak belongs to;
(2) We integrated Craack’s output with NVR-BIP;
(3) We simulated the HADAMAC experiment to predict the amino acid class that each NMR peak belongs to;
(4) We incorporated the HADAMAC experiment into NVR-BIP; and
(5) We tested our approach on NVR-BIP’s data set and compared our results with NVR-BIP.
The rest of the paper is organized as follows: In Section 1, we review the previous approaches. The incorporation of amino acid type predictions made by Craack into NVR is described in Section 2. In Section 3, we review the HADAMAC experiment and integration of HADAMAC experiment with NVR-BIP. Data preparation is in Section 4 and the experimental results are in Section 5. We conclude and discuss future work in Section 6.
In NMR assignments, the problem is to find the correspondence between a set P of peaks and a set A of residues. A scoring function determines the score associated with assigning each NMR peak p to each amino acid a. The scoring functions in SBA makes use of the template structure to compute this function. Due to the errors and noise in experimental data, and the assumptions made in developing a scoring function (such as Gaussian assumption for the distribution of data), the assignment having the optimum score may not be the 100% correct assignment.
NVR
NVR-BIP uses the Nuclear Vector Replacement (NVR) framework [5,6], and incorporates additional sources of data, to determine the assignments. The type of data accepted by NVR-BIP is as follows:
1. Chemical shifts for 15N and HN atoms
2. Unambiguous backbone NOEs
3. TOCSY data if available
4. RDCs if available
5. Hydrogen-Deuterium exchange data if available.
These data sources (except the NOEs) are combined into a scoring function where lower scores are associated with more likely assignments. If the assignment probability is very small, the score associated with the corresponding assignment is +∞.
NVR-BIP formulates the problem as a binary integer program where the objective is to find the assignment whose total score is minimum subject to the NOE constraints. NVR-BIP uses a BIP solver to find the minimum scoring assignment. NVR-BIP was tested on 7 proteins with 25 templates and resulted in higher accuracies than NVR-EM [4,6].
Amino acid typing
Amino acid typing involves identifying the type of an amino acid based on NMR data such as chemical shifts. Example programs for amino acid typing include TATAPRO II [9], which takes in CA and CB chemical shifts and outputs one out of 8 categories to which the amino acid may belong to. Alternative to typing is the HADAMAC [8] experiment which enables to successfully distinguish the type of the amino acid in about half an hour.
Craack [7] is a tool that takes chemical shifts {N,HN,HA, HB,CA,CB,CO} as input and outputs a list of amino acid types. Each predicted amino acid type has a confidence score. Craack uses different amino acid type prediction tools such as Rescue [10], RescueN [11], Rescue2 [12], Platon [13], and SVMTyping [7]. Craack gets the prediction values of these tools and uses two approaches to compute a single consensus score value for the amino acid type corresponding to the chemical shift values. In the first approach, the amino acid types are categorized into eight groups and support vector machines (SVM) are used to determine the confidence score of the amino acid group. In the second approach, the consensus score is computed by voting in which each source (e.g. the aforementioned prediction tools and consensus score of SVM) has experimentally pre-determined weights. We used the consensus scores in our experiments, which gives the predictions for each amino acid separately.
Amino acid typing has been incorporated previously into NMR SBA. The approach of [14] utilizes Rescue [10] software which is an earlier work by the authors of Craack [7]. They utilize experimental chemical shifts from the BMRB [15] that they feed into the Rescue software. The approach is tested on synthetic data from five proteins with assignment accuracies ranging between 77-100% when the errors in amino acid typing are not corrected. Another approach that also uses Rescue is [16], which has been tested on three proteins with up to 166 residues and an assignment accuracy varying between 60% and 80%. Our approach utilizes Craack which uses Rescue and four other amino acid typing tools and takes the consensus of their predictions. We also utilize HADAMAC which to our knowledge has not been used before for NMR SBA. Furthermore, HADAMAC experiment has the advantage of being practically error free on relatively smaller proteins.
The main motivation of this work is to investigate whether amino acid typing can be used to improve the accuracy of NVR-BIP. To that end, we provide chemical shifts to Craack and obtain amino acid predictions along with confidence scores. This results in a matrix (Craack score) that has for each (peak, residue) pair the consensus score associated by Craack. We integrate this matrix with NVR’s score matrix using two approaches [17].
Our notations for the score matrices is as follows: Let Sn be the scoring matrix of NVR and Sc be the scoring matrix of Craack. Then, Sn[i][j] = sn corresponds to the NVR score of assigning peak i to amino acid j. The lower this value, the higher is the probability of assignment according to NVR. Similarly, Sc[i][j] = sc corresponds to Craack score of assigning peak i to amino acid j. Unlike Sn, this value is proportional to the assignment probability according to Craack. Sn is equal to ∞ if the assignment of peak i to residue j is impossible according to NVR. Sc is 0 if amino acid j is not among the list of residues returned by Craack.
Only pruning amino acid candidates with Craack
This approach uses Craack as a filter to eliminate the possibility of certain assignments. If the type of the considered residue is not amongst the set of amino acid possibilities returned by Craack, the corresponding score is assigned an infinite value and that assignment possibility is eliminated. More formally, for each peak i and for each amino acid j the combined score matrix that is derived from this approach (S1nc) is defined as follows:
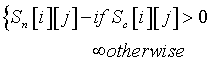
Pruning + rewarding amino acid candidate predictions of Craack
The idea of this approach is to reward the assignments whose Craack score is positive. Therefore, we subtract Craack score from NVR score. But if the Craack score is 0 then the corresponding assignment possibility is eliminated. More formally, for each peak i and for each amino acid j the combined score matrix that is derived from this approach (S2nc) is defined as follows:
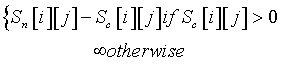
HADAMAC [8] experiment uses Hadamard encoded amino acid type editing scheme. In Hadamard encoded type editting, first, the twenty amino acids are grouped into seven classes. The different classes correspond to Gly (1), Val, Ile (2), Ala (3), Thr (4), Asn, Asp (5), Phe, Tyr, Trp, His, Cys, Ser (6), and Arg, Glu, Lys, Pro, Gln, Met and Leu (7) side chains. Then each peak is assigned to one of these seven classes which represents the type of the previous residue of the residue corresponding to the peak.
We simulate the HADAMAC experiment [18]. We assign each peak i to one of the seven classes according to the type of the residue j−1, where j is the residue that is to be assigned to peak i. We use H(i) to represent the set that contains the amino acid types corresponding to peak i according to the HADAMAC experiment, and we use typej to represent the type of the residue j. Given the NVR scoring function Sn(i,j) which is defined for each peak-residue pair, we compute the new scoring function, Snh, using the HADAMAC experiment as follows:
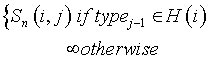
This new scoring function is similar to NVR’s scoring function where some of the peak-residue assignments are pruned.
We test our approach on the data set of NVR-BIP using the chemical shifts collected from various sources. NVR-BIP only requires 15N and HN chemical shifts. Although Craack can run with this minimal set of data, the predictions are not accurate. Therefore we provided Craack with the full list of chemical shifts. We predicted this data using SHIFTS [19] and SHIFTX [20]. For some proteins we also used experimental chemical shifts collected from BMRB [15]. We have tested our approach on NVR-BIP’s test set in order to compare the results. The proteins we have tested our approach on are: ubiquitin (template pdb ids: 1UBI, 1UBQ, 1G6J, 1UD7, 1AAR), streptococcal protein G (template pdb ids: 1GB1, 2GB1, 1PGB), lysozyme proteins (template pdb ids: 193L, 1AKI, 1AZF, 1BGI, 1H87, 1LSC, 1LSE, 2LYZ, 3LYZ, 4LYZ, 5LYZ, 6LYZ), human Set 2-Rpb1 interacting domain (hSRI), the FF Domain 2 of human transcription elongation factor CA150 (RNA polymerase II C-terminal domain interacting protein) (ff2), Y-polymerase Eta (pol η), B1 domain of streptococcal protein G (GB1).
We performed experiments to compare the results of NVR+Craack and NVR+HADAMAC on the dataset of NVR-BIP mentioned in Section 5. This test set was constructed by including to the set of proteins on which the original NVR approach was tested [6] additional proteins for which NMR data was collected by the group of Prof. Zhou from Duke University [4].
Tables 1, 2, 3, and 4 show the results of the experiments. With NVR+Craack, the assignment accuracies improved by up to 15% with only pruning. On the other hand, the assignment accuracies improved by up to 21% with the approach that also rewards Craack predictions. The only exceptions are 4LYZ and 5LYZ for which the accuracies of the assignments of NVR-BIP were 91% but they decreased by 4% when RDCs are available. NVR+HADAMAC consistently outperformed NVR-BIP. The assignment accuracies improved by up to 21% when we used NVR+HADAMAC instead of NVR-BIP. For most cases, NVR+HADAMAC resulted in higher accuracies than NVR+Craack. The assignment accuracies improved by up to 17% when we use NVR+HADAMAC instead of NVR+Craack. The reason for this superior performance is that Craack makes a computational prediction by taking the consensus of multiple amino acid type prediction tools and is prone to error (which also explains its lower performance for 4LYZ and 5LYZ mentioned above), whereas HADAMAC simply classifies each peak into one of the amino acid groups. The low resolution provided by HADAMAC is compensated by its practically error-free data.
| Protein | RDCs | NVR-BIP [4] | NVR+CRAACK (Section 3.1) | NVR+CRAACK (Section 3.2) | NVR+HADAMAC |
| 1UBI | without RDCs with RDCs |
87% 100% |
97% 100% |
97% 100% |
96% 100% |
| 1UBQ | without RDCs with RDCs |
87% 100% |
97% 100% |
100% 100% |
96% 100% |
| 1G6J | without RDCs with RDCs |
87% 93% |
93% 93% |
97% 100% |
91% 96% |
| 1UD7 | without RDCs with RDCs |
81% 97% |
87% 97% |
90% 97% |
90% 99% |
| 1AAR | without RDCs with RDCs |
79% 100% |
94% 100% |
100% 100% |
96% 100% |
Table 1: Results on ubiquitin.
| Protein | RDCs | NVR-BIP [4] | NVR+CRAACK (Section 3.1) | NVR+CRAACK (Section 3.2) | NVR+HADAMAC |
| 1GB1 | Without RDCs with RDCs |
100% 100% |
100% 100% |
100% 100% |
100% 100% |
| 2GB1 | Without RDCs with RDCs | 100% 100% |
100% 100% |
100% 100% |
100% 100% |
| 1PGB | Without RDCs with RDCs | 96% 100% |
96% 100% |
96% 100% |
100% 100% |
Table 2: Results on streptoccocal protein G.
| Protein | RDCs | NVR-BIP [4] | NVR+CRAACK (Section 3.1) | NVR+CRAACK (Section 3.2) | NVR+HADAMAC |
| 193L | Without RDCs with RDCs |
78% 100% |
79% 100% |
79% 100% |
95% 100% |
| 1AKI | Without RDCs with RDCs | 78% 98% |
80% 98% |
80% 98% |
93% 98% |
| 1AZF | Without RDCs with RDCs | 74% 94% |
76% 95% |
78% 95% |
95% 95% |
| 1BGI | Without RDCs with RDCs |
75% 97% |
79% 97% |
83% 97% |
95% 100% |
| 1H87 | Without RDCs with RDCs | 77% 100% |
79% 100% |
79% 100% |
95% 100% |
| 1LSC | Without RDCs with RDCs | 74% 100% |
78% 100% |
79% 100% |
95% 100% |
| 1LSE | Without RDCs with RDCs |
75% 98% |
78% 98% |
79% 98% |
95% 98% |
| 1LYZ | Without RDCs with RDCs | 79% 82% |
81% 87% |
79% 87% |
95% 95% |
| 2LYZ | Without RDCs with RDCs | 75% 91% |
79% 95% |
79% 95% |
95% 97% |
| 3LYZ | Without RDCs with RDCs |
79% 90% |
83% 90% |
83% 90% |
95% 97% |
| 4LYZ | Without RDCs with RDCs | 75% 91% |
79% 87% |
79% 87% |
95% 97% |
| 5LYZ | Without RDCs with RDCs | 75% 91% |
79% 87% |
79% 87% |
95% 97% |
| 6LYZ | Without RDCs with RDCs |
75% 96% |
79% 97% |
81% 97% |
95% 100% |
Table 3: Results on lysozyme.
| Protein | RDCs | NVR-BIP [4] | NVR+CRAACK (Section 3.1) | NVR+CRAACK (Section 3.2) | NVR+HADAMAC |
| ff2 | Without RDCs with RDCs |
%85 %93 |
%93 %93 |
%93 %93 |
%92 %98 |
| hSRI | Without RDCs with RDCs | %73 %89 |
%73 %89 |
%81 %94 |
%88 %97 |
| pol | Without RDCs with RDCs |
%100 %100 | %100 %100 |
%100 %100 |
%100 %100 |
| GB1 | Without RDCs with RDCs | %96 %100 |
%100 %100 |
%100 %100 |
%100 %100 |
Table 4: Results on ff2, hSRI, pol η and GB1.
This paper investigated using two different types of NMR data: chemical shifts for additional atoms or the HADAMAC experiment in the NVR framework. In the former approach, additional chemical shifts enabled the use of an amino acid type prediction tool (Craack) which helped improve NVR’s scoring function. This approach pruned those amino acids which were not in the list of amino acid candidates predicted by Craack, and considered using the Craack score to modify NVR’s scoring function for the remaining amino acid candidates. The latter approach used HADAMAC experiment which was only used to prune the space of possible assignments.
Our use of amino acid typing in NVR is similar to the approach of [14] as both methods used amino acid typing to help the assignment process, however our approach used Craack tool (which supersedes the Rescue tool used in [14]) and HADAMAC experiment which have to the best of our knowledge not been considered before. The use of amino acid typing was previously implicit in NVR’s scoring function with the incorporation of TOCSY data, however TOCSY data may not be available for some proteins. This approach provides an alternative method for such proteins and augments NVR’s performance on the test set for which TOCSY data is also available. Our approach suggested that it is possible to improve NVR-BIP’s assignment accuracy by incorporating these additional types of data. This work is also a followup to [4] where the contribution of each of the data sources into NVR’s accuracy was studied.
Note that the existing approaches to NMR SBA use different types of NMR data. For instance, the approach of [16] use 3JHNHα data in addition to those used by NVR, the NOEnet [21] approach uses only 1HN-1HN unambiguous NOEs, and the approach in [14] use both ambiguous and unambiguous NOEs. Therefore it is not possible to directly compare our accuracies with other NMR SBA software.
There are various amino acid groupings in the literature, such as [22,23]. The amino acid groupings mentioned in this work come from external constraints - the Craack approach can classify each amino acid into eight classes and similarly HADAMAC classifies each amino acid into one of seven classes. We use the Craack version that issues a classification for each amino acid type separately, and HADAMAC groupings are imposed by the experiment.
Note that NVR-BIP’s test set is entirely separate from the training set of SHIFTS and SHIFTX, except 1UBQ which is used in the training set of SHIFTX. 1UBQ was in the test set of NVR in previous versions [4,6] and was retained for consistency purposes. Craack has been trained and tested on chemical shifts from BMRB; since BMRB represents experimental chemical shifts, our approach also uses BMRB chemical shifts as input. Furthermore, HADAMAC method refers to an experiment and does not involve a training set.
The results indicate that the approaches proposed in Section 3 are potentially useful for SBA since in general they lead to better assignment accuracies. Although our proposal for combining NVR score with Craack in Section 3.2 is simple, it lead to improvements in assignment accuracies. As future work, it may be possible to normalize the NVR score and Craack score before combining them. It may also be possible to tolerate the incorrect predictions of Craack by iteratively performing the assignments as in [14], first with the amino acid typing strictly enforced, fixing some assignments, and then relaxing the type matching requirement. This may make our tool more robust with respect to errors in chemical shifts.
We also proposed an approach to integrate HADAMAC experiment with NVR’s data types. The experimental results shown in the previous section indicate that the proposed approach leads to better accuracies than NVR-BIP and NVR+Craack. With the addition of the HADAMAC experiment, NVR becomes a more useful and practical tool that can be used in an NMR laboratory. Furthermore, HADAMAC experiment distinguishes the type of the amino acid in about 30 minutes; whereas conventional 3D experiments needed to acquire the data used by Craack take hours to complete.
1) On the other hand, HADAMAC experiment has some limits. These are as followIn order to measure HADAMAC data, we need to have reasonably well resolved HSQC crosspeaks. There can be partially overlapping peaks but there will be trouble for exactly overlapped 2D crosspeaks.
2) HADAMAC works well only for reasonably small proteins (up to about 15kDa)
3) The protein needs to be fully protonated, at least for the beta position.
4) The protein has to be 13C and 15N labeled.
5) HADAMAC experiment does not provide information for the last residue in protein sequence and for residues preceding proline residues since they are not followed by a residue with the HN moiety.
Note that the experiments were performed on theoretical HADAMAC data except for ubiquitin. Furthermore, our approach is tested on manually picked peaks, as extracted from BMRB. An area of future work is to make the tool more useful for the NMR spectroscopist by utilizing automatically picked peaks. Another area of future study is to incorporate additional types of real data into NVR, such as ambiguous NOEs, and use the intensity field of the NOEs to perform the assignments.
The source code of the software is available upon request.
We thank Dr. Ewen Lescop for discussions. This work was supported by following grants to M.S.A.: The Scientific and Technical Research Council of Turkey research support program (program code 1001) [109E027] and EU Marie Curie Grant PIRG05-GA-2009-249267.