Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2015) Volume 4, Issue 4
Pancreatic ductal adenocarcinoma (PDAC) has a very poor prognosis. Meaningful therapeutic options for unresectable PDAC remain limited and therefore more efficient therapeutic strategies are urgently required. We explored the approach of manipulating dose intensity of cytotoxic agents through prolonged therapy and by combination with antiangiogenic agents in experimental PDAC. Median animal survival over controls (19 days) was increased after prolonged therapy (up to 6 weeks) with gemcitabine to 29 days (a 53% increase, p = 0.008). Addition of two antiangiogenic agents, bevacizumab and EMAP, further extended median survival to 46 days (a 142% increase, p = 0.0001). Corresponding survival extension after therapy with similar agents limited to 2 weeks was 37% for gemcitabine and 127% after gemcitabine plus antiangiogenic agents. Pharmacokinetic analysis revealed that the addition of antiangiogenic agents caused a 4.4-fold increase in gemcitabine plasma concentration and 8-fold increase in tumor concentration compared with gemcitabine monotherapy group. Prolonged therapy of docetaxel also increased animal survival (38 days, a 100% increase, p = 0.0004) that was further extended by the addition of the antiangiogenic combination (49 days, a 158% increase, p = 0.0001). In vitro evaluation of exposure time effects on cell proliferation revealed that in PDAC cells (AsPC-1) longer exposure (120 h) of gemcitabine or docetaxel caused greater relative inhibition in cell proliferation compared with 72 h exposure. At 1ïM concentration, inhibition in cell proliferation after 72 h and 120 h was 27% and 55% (gemcitabine); 64% and 84% (docetaxel). Only in endothelial cells and fibroblasts but not in PDAC cells did the combination with antiangiogenic agents create additive effects over each cytotoxic agent alone. These findings demonstrate that increasing bioavailability of cytotoxic agents, both via increased exposure through prolonged therapy or greater intratumoral dose intensity by combination with antiangiogenic agents could enhance in vivo antitumor efficacy and thus warrants further investigation.
<Keywords: Pancreatic cancer; Gemcitabine; Docetaxel; Bevacizumab; EMAP; Chemotherapy
PDAC: Pancreatic Ductal Adenocarcinoma; Gem: Gemcitabine; DT: Docetaxel; EMAP or E: Endothelial Monocyte Activating Polypeptide; Bev: Bevacizumab
Pancreatic ductal adenocarcinoma (PDAC) ranks the fourth most common cause of all cancer deaths in the United States, and is expected to become the second-leading cause of cancer death by 2030 [1]. The prognosis of patients with advanced PDAC remains very poor with a median survival of 4 to 6 months; for all stages combined, the 5-year survival rate is only 6% [2]. At the time of diagnosis, about 80% of PDAC patients are unresectable due to distant metastasis or locoregional extent [3,4]. Furthermore, in those 10-20% of patients who present with localized disease and undergo surgical resection, rapid distant or locoregional relapse is very common [4,5]. Therapeutic options for unresectable PDAC are limited, and single agent gemcitabine has been the standard of care since 1997 until recently [6]. Taxanes, docetaxel and paclitaxel, showed promising antitumor activity in other solid tumors [7,8] but showed dismal activity in PDAC [9,10]. Recently, greater efficacy against advanced pancreatic cancer has been demonstrated through the combination of gemcitabine with nabpaclitaxel [11] or of multiple cytotoxic agents in form of FOLFIRINOX [12,13]. Despite these recent trial-based incremental improvements in patient outcome, prognosis of PDAC patients still remains dismal.
Therefore, novel and more efficient therapeutic strategies to prolong survival of PDAC patients require immediate attention.
Angiogenesis is an essential and crucial step in the development and progression of cancer including PDAC. Among several positive regulators of angiogenesis, vascular endothelial growth factor (VEGF), a multifunctional cytokine, is one of the most powerful regulators of tumor vasculature. VEGF not only promotes angiogenesis by inducing proliferation, migration, and survival of endothelial cells [14], but also increases vessel permeability [15]. VEGF is overexpressed in pancreatic cancer [16], correlates with local disease progression [17] and poor prognosis [18], providing a strong rationale for exploiting the therapeutic potential of anti-VEGF agent combinations with an effective cytotoxic regimen. Bevacizumab, a recombinant humanized monoclonal antibody against VEGF, was evaluated in PDAC clinical studies. Although the bevacizumab and gemcitabine combination showed some promise in a phase II trial, no significant benefit was observed in subsequent phase III studies [19]. Endothelial monocyte activating polypeptide II (EMAP) is a proinflammatory cytokine with antiangiogenic and anti-endothelial activities. EMAP has potent effects on endothelial cells such as inhibition of proliferation, migration, and induction in apoptosis [20,21]. EMAP suppresses primary and metastatic tumor growth [20,22,23] that so far could be linked in part to its inhibitory activity on VEGF and fibronectin signaling [24,25]. EMAP has also been shown to improve gemcitabine and docetaxel antitumor response [26-28]. Furthermore, in PDAC animal survival studies, addition of two antiangiogenic agents, bevacizumab and EMAP, to gemcitabine was more effective than single agent addition, and a prolonged therapy group with this most effective combination had some benefit over short term therapy [26]. In the present study, we examined the antitumor potential for increasing bioavailability of cytotoxic agents, either via prolonged therapy or by addition of polymechanistic anti-endothelial therapy with bevacizumab + EMAP, within experimental pancreatic cancer.
Materials
Gemcitabine was purchased from Eli Lilly (Indianapolis, IN). Docetaxel (Taxotere) was purchased from Sanofi-Aventis (Bridgewater, NJ). Bevacizumab was purchased from Genentech (South San Francisco, CA). Recombinant human EMAP was prepared and purified in the lab as previously described [29], and the cell proliferation reagent WST-1 was purchased from Roche Diagnostic Corporation (Indianapolis, IN).
Cell culture
The human pancreatic cancer cell line AsPC-1 and human fibroblast cell line WI-38 was purchased from the American Type Culture Collection (Rockville, MD). AsPC-1 and WI-38 cells were grown in RPMI 1640 medium and DMEM, respectively (Sigma Chemical Co. St. Louis, MO) supplemented with 10% fetal bovine serum. Human umbilical vein endothelial cells (HUVECs) were purchased from Lifeline Cell Technology (Walkersville, MD) and were grown in VascuLife medium containing endothelial cell growth supplements.
Cell viability assay
In vitro cell proliferation assays were performed in 96-well plates using the colorimetric WST-1 reagent. Four thousand cells were plated in a 96-well plate and after 16 hours the medium was replaced with low serum medium. Cells were treated with gemcitabine, docetaxel, bevacizumab and EMAP. The range of concentrations used for gemcitabine, docetaxel and EMAP were from 50 nM to 10 μM; and for bevacizumab 10 μg/ml to 1 mg/ml. After 72 h or 120 h incubation, WST-1 reagent (10 μl) was added in each well and absorbance was measured at 450 nm after 2 hours using a microplate reader. Drug sensitivity graphs were plotted using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA).
Subcutaneous xenografts and pharmacokinetic analysis
All animals were housed in a pathogen-free facility and had access to food and water ad libitum. Animal experiments were performed according to the guidelines and approved Institutional Animal Care and Use Committee protocols of the University of Texas Southwestern Medical Center (Dallas, TX; protocol number 2008-0348) and the Indiana University School of Medicine (South Bend, IN; protocol number 16-023). Athymic female nu/nu mice (4-6 weeks) were used to establish the subcutaneous xenograft model. Two weeks after subcutaneous injection of 0.75 x 106 AsPC-1 cells, mice were randomly grouped and treated intraperitoneally with PBS (control), gemcitabine (100 mg/kg, twice weekly), bevacizumab (10 μg/mouse, twice weekly) or EMAP (80 μg/kg, 5 times per week) for two weeks. The tumor size was measured twice weekly and tumor volume (V) was calculated by using the formula [V = ½ (L × (W) 2], where L = length and W = width of the tumor mass.
Gemcitabine concentrations in tumor and plasma were analyzed as previously described [30]. Tumor harvesting and plasma collection were performed at a time point that was 24 hours after the final bevacizumab or EMAP treatment and 4 hours after the final gemcitabine treatment. Tumor samples, snap frozen in liquid N2, were homogenized in PBS containing 100 μg/ml tetrahydrouridine (THU) to prevent ex vivo metabolism of gemcitabine (2’, 2’-difluoro-2’-deoxycytidine, dFdC) to the inactive metabolite, 2’-2’-difluorodeoxyuridine (dFdU). Plasma collection was also performed in the presence of 100 μg/ml of THU. Analytical methods were developed to detect gemcitabine using an Applied Biosystems / MDS Sciex 4000 QTRAP mass spectrometer coupled to a Shimadzu Prominence LC. Chromatography was performed using a Phenomenex Luna C8 column. Pharmacokinetic analysis was performed using the Phoenix® WinNonLin® (Certara L.P., Princeton, NJ) software package. Non-compartmental modeling with sparse sampling was utilized.
Animal survival analysis
Animal survival studies were performed using 6 to 8 week old female NOD/SCID mice, as previously described [31]. Mice were intraperitoneally injected with AsPC-1 cells (0.75 × 106) and were randomly grouped (n = 6 to 8 per group) after two weeks. They were then treated intraperitoneally for two weeks (standard, short-term therapy) or up to six weeks (prolonged therapy) with PBS (control), gemcitabine (100 mg/kg, twice per week), docetaxel (4 mg/kg, twice per week), bevacizumab (10 μg/mouse, twice per week) or EMAP (80 μg/ kg, 5 times per week) or until the need to discontinue treatment earlier. In these cases, animals were euthanized when apparently moribund according to predefined criteria including rapid body weight gain or loss (>15%), tumor size, lethargy, inability to remain upright and lack of strength. Animal survival was evaluated from the start of therapy until death.
In vitro cell proliferation assay results are expressed as mean ± standard deviation (SD). Statistical significance was analyzed by the two-tailed Student’s t-test using GraphPad Prism 6.0 Software. Significant differences in animal survival between groups were determined by log-rank testing using GraphPad Prism 6.0 Software. P values of <0.05 were considered to represent statistically significant group differences.
Animal survival extension by prolonged gemcitabine therapy and addition of antiangiogenic agents
Human intraperitoneal xenograft studies in NOD/SCID mice resulted in a median survival of 19 days in the PBS treated control group. Relative to controls, median survival was significantly increased by a prolonged therapy of gemcitabine (29 days, a 53% increase, p = 0.008). There was a minor survival benefit after prolonged therapy with antiangiogenic agents alone (22 days, a 16% increase, p = 0.02). Gemcitabine-mediated animal survival was further extended by the addition of antiangiogenic agents (46 days, a 142% increase, p = 0.0001; Figure 1A). The relative increase in median animal survival by prolonged therapy was significantly higher than the corresponding median survival extension after standard therapy with similar agents limited to 2 weeks; in this case, the increase in survival was 37% for gemcitabine and 127% after gemcitabine plus antiangiogenic agents, respectively (Figure 1B). There were no apparent toxicity-related signals in terms of rapid animal weight loss or gain (>15%), general appearance, habitus and activity levels, during the therapy period until approximately a week before animal death.

Two weeks after intraperitoneal injection of AsPC-1 cells into NOD/SCID mice, treatment was given either for 2 weeks with gemcitabine (100 mg/kg, twice per week), bevacizumab (10 μg/mouse, twice per week) or EMAP (80 μg/kg, 5 times per week) or with gemcitabine (100 mg/kg, twice per week), bevacizumab (10 μg/mouse, twice per week) or EMAP (80 μg/kg, 5 times per week) for up to next six weeks. (A) The graph represents the animal survival time from the beginning of therapy. Statistical group differences in survival time were calculated using log rank testing (GraphPad Prism 6.0). (B) Comparison on percent increase in survival after standard therapy and prolonged therapy. Asterisk (*) represents percent increase in animal survival after 2 weeks treatment as previously published from our lab and referenced in the results section. (C) Gemcitabine concentrations in snap frozen tumor sections or in the plasma were analyzed as described in the methods section and plotted as bar graph. The data are expressed as the mean ± standard deviation (n=3).
Figure 1: Prolonged therapy effects of gemcitabine and addition of anti-angiogenic agents on animal survival.
Gemcitabine concentration in tumor and plasma: effect of addition of antiangiogenic agents
Evaluation of the mechanism by which antiangiogenic agents may enhance the gemcitabine antitumor response revealed that addition of antiangiogenic agents increased gemcitabine concentration in tumor and plasma (Figure 1C). Addition of antiangiogenic agents caused a 4.4 fold increase in gemcitabine plasma concentration compared with the gemcitabine monotherapy group. Furthermore, addition of antiangiogenic agents also caused an 8-fold increase in gemcitabine tumor concentrations compared with the gemcitabine monotherapy group (Figure 1C). Furthermore, we observed that corresponding with the increase in gemcitabine concentration in plasma and tumor by addition of antiangiogenic agents, the same antiangiogenic agents created additive effects on gemcitabine-mediated local tumor growth inhibition (data not shown).
Animal survival benefits by prolonged docetaxel therapy: Addition of antiangiogenic agents
Evaluation of animal survival benefit by prolonged therapy with docetaxel revealed that compared to controls (19 days) docetaxel therapy increased animal survival to 38 days (a 100% increase, p = 0.0004) (Figure 2). The improvement in animal survival through prolonged docetaxel therapy was greater than the 75% increase after standard two-week docetaxel therapy as reported earlier [28]. Prolonged docetaxel therapy-related animal survival was further extended by the addition of antiangiogenic agents to 49 days (a 158% increase, p = 0.0001) (Figure 2). Again, there were no apparent toxicity related signals in terms of rapid animal weight loss or gain (>15%), general appearance, habitus and activity levels, during the therapy period until approximately a week before animal death.

AsPC-1 cells were injected intraperitoneally into NOD/SCID mice and treatment started after 2 weeks with docetaxel (4 mg/kg, twice per week), bevacizumab (10 μg/mouse, twice per week) or EMAP (80 μg/kg, 5 times per week). The graph represents the animal survival time from the beginning of therapy. Statistical group differences in survival time were calculated using log rank testing (GraphPad Prism 6.0).
Figure 2: Prolonged therapy effects of docetaxel and addition of anti-angiogenic agents on animal survival.
Effect of chemotherapy exposure time and addition of antiangiogenic agents on PDAC cell in vitro proliferation
In vitro cell proliferation analysis in AsPC-1 cells exposed to gemcitabine showed a significant dose- and exposure timedependent inhibition in cell proliferation (Figure 3). At 1 μM and 10 μM gemcitabine, inhibition in cell proliferation was 27% and 44% after 72 h of drug exposure, respectively. With similar gemcitabine concentrations, this inhibition was 55% and 69% after 120 h of drug exposure (Figure 3A). Single agent bevacizumab or EMAP had no significant inhibitory effect on AsPC-1 cell proliferation (Figure 3B). Furthermore, combination of gemcitabine with antiangiogenic agents had no significant additive effects after 72 h or 120 h of drug exposure (Figure 3B).
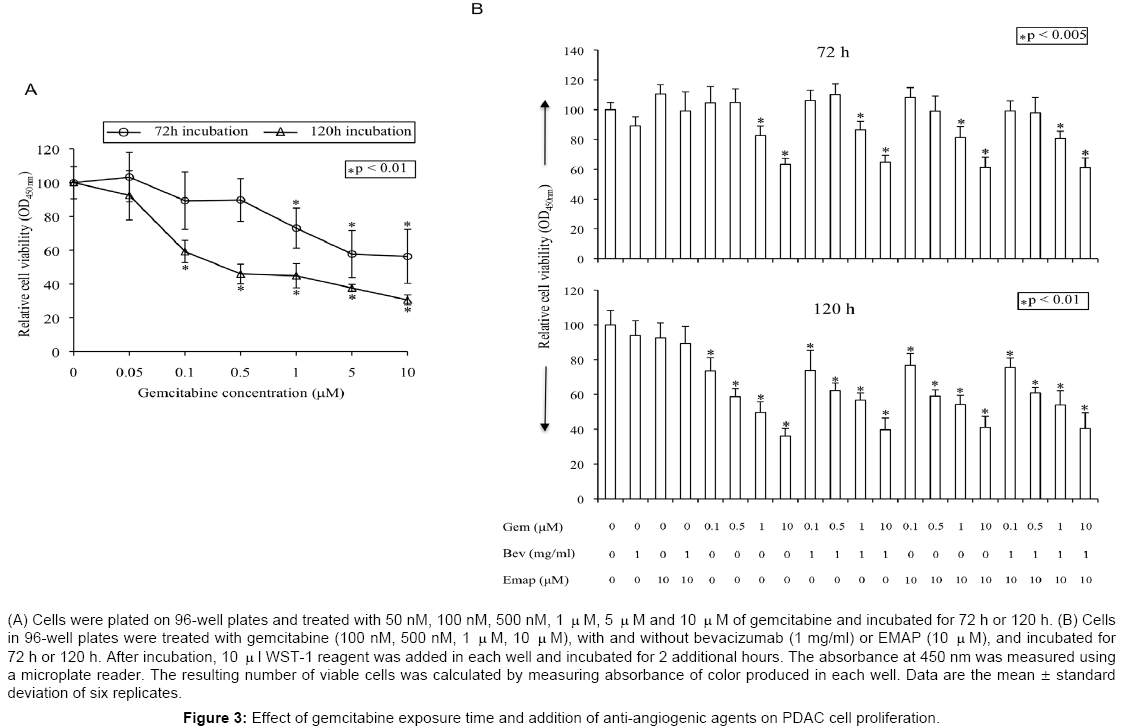
Figure 3: Effect of gemcitabine exposure time and addition of anti-angiogenic agents on PDAC cell proliferation.
Similarly, at 1 μM and 10 μM docetaxel, inhibition in cell proliferation was 64% and 67% after 72 h of drug exposure, compared to 83% and 86% after 120 h of drug exposure (Figure 4A). Again, single agent bevacizumab and EMAP showed no significant inhibitory effect on AsPC-1 cell proliferation, and the combination of docetaxel with antiangiogenic agents had no significant additive effects after 72 h or 120 h of drug exposure (Figure 4B).

Figure 4: Effect of docetaxel exposure time and addition of anti-angiogenic agents on PDAC cell proliferation.
Effect of chemotherapy exposure time and addition of antiangiogenic agents on endothelial cell and fibroblast proliferation
In HUVECs, 1 μM and 10 μM concentrations of gemcitabine caused 75% and 79% inhibition in cell proliferation after 72 h of drug exposure. Single agent bevacizumab at 500 μg/ml and 1 mg/ml concentrations caused 13% and 22% inhibition in cell proliferation after 72 h of drug exposure. Single agent EMAP at 5μM and 10 μM caused 14% and 22% inhibition in cell proliferation after 72 h of exposure (Figure 5). Combination of gemcitabine with antiangiogenic agents resulted in additive effects on proliferation inhibition. Combination treatment of gemcitabine (1 μM) with bevacizumab (500 μg/ml), EMAP (5 μM) or bevacizumab + EMAP caused 77%, 80% and 86% inhibition in cell proliferation (Figure 5). In HUVECs, effect of 120 h exposure time of gemcitabine and combination of antiangiogenic agents at similar concentrations could not be evaluated in part due to significant cell death in the control group without drug treatment.
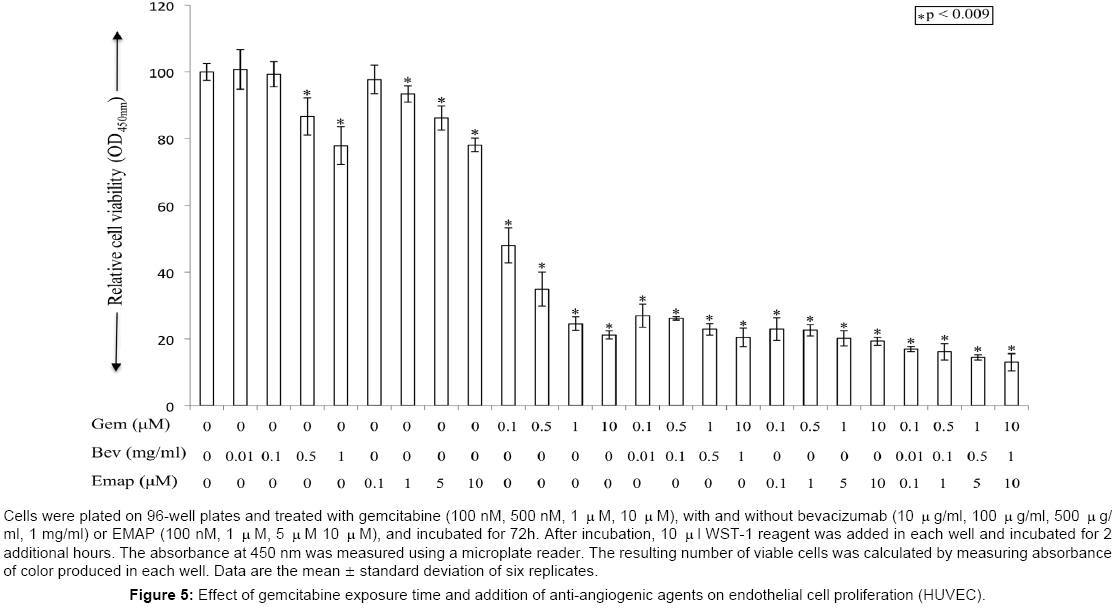
Figure 5: Effect of gemcitabine exposure time and addition of anti-angiogenic agents on endothelial cell proliferation (HUVEC).
In WI-38 fibroblast cells, gemcitabine concentrations 1 μM and 10 μM caused 70% and 77% inhibition in cell proliferation after 72 h of drug exposure. Single agent bevacizumab at 500 μg/ml and 1 mg/ ml concentrations caused 12% and 29% inhibition in cell proliferation after 72 h drug exposure. Single agent EMAP at 5 μM and 10 μM caused 17% and 27% inhibition in cell proliferation after 72 h exposure (Figure 6). Combination of gemcitabine with antiangiogenic agents resulted in additive effects on proliferation inhibition, since 1 μM gemcitabine together with bevacizumab at 500 μg/ml, EMAP at 5 μM or bevacizumab + EMAP caused 72%, 78% and 88% inhibition in cell proliferation (Figure 6). Similar to HUVECS, in WI-38 cells the effect of 120 h exposure of gemcitabine in combination with antiangiogenic agents could not be evaluated due to significant cell death in notreatment control group.
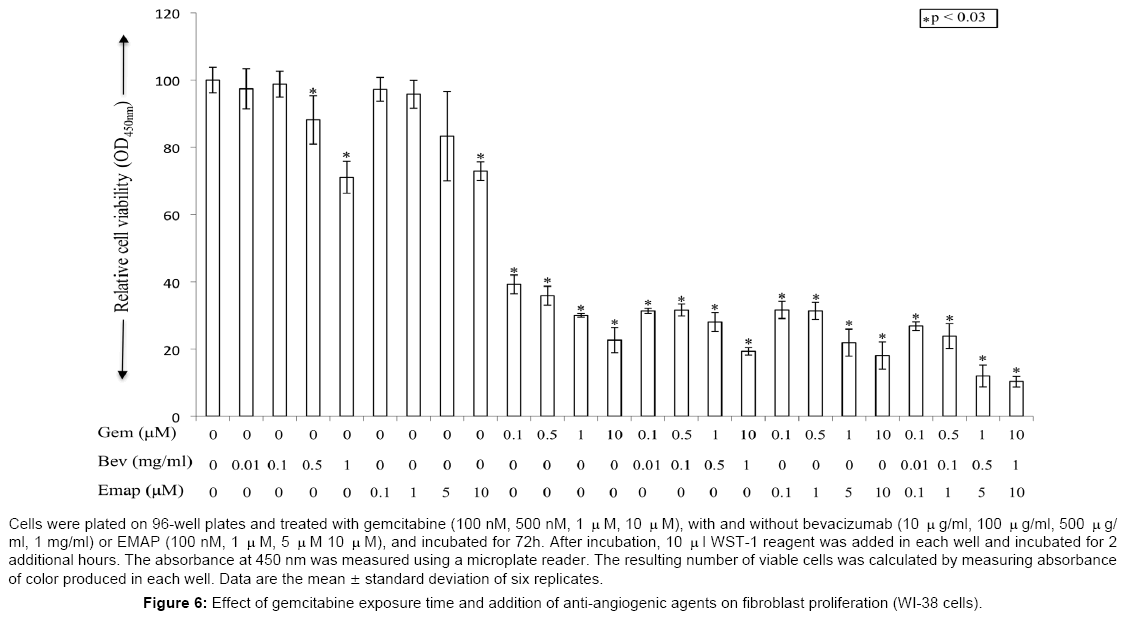
Figure 6: Effect of gemcitabine exposure time and addition of anti-angiogenic agents on fibroblast proliferation (WI-38 cells).
Pancreatic cancer is characterized by rapid progression, invasiveness and marked resistance to conventional chemotherapy. Systemic chemotherapy remains the only therapeutic option for most patients either due to unresectable disease at diagnosis or recurrence after pancreatectomy. Since current cytotoxic chemotherapy options remain fairly ineffective, it is important to develop novel strategies to enhance the tumor response to systemic therapy. The scope of the present study was to evaluate the effectiveness of altering dose intensity of cytotoxic agents, either in form of greater exposure over time through prolonged therapy, or by addition of antiangiogenic agents through their possible impact on cytotoxic agent delivery into the tumor.
The present study results show the antitumor efficacy of prolonged therapy of both gemcitabine and docetaxel to a greater level than 2-week therapy in an intraperitoneal xenograft survival model that is well representative of the progression pattern of PDAC [26,28,32]. Importantly, prolonged therapy did not cause any obvious loss of efficacy over time, but appeared to lead to additional relative therapeutic gains for the combination treatment groups. In the model setting used, no evidence for loss of antitumor effects with prolonged therapy due to development of resistance was thus obtained.
Molecular-targeted agents may potentially contribute to improvement in long-term disease control, and many of these agents are better tolerated than traditional cytotoxic agents. Targeting tumor angiogenesis through a polymechanistic approach is an attractive strategy for PDAC therapy because of their potential for synergistic interaction with cytotoxic agents, limited toxicity and enhanced antitumor effects [33]. Previous studies in our lab have shown the benefits of combining several multitargeting antiangiogenic agents with cytotoxic chemotherapy for PDAC therapy [26,28,32]. In the present study, prolonged therapy with antiangiogenic agents alone generated only a small effect on improving animal survival, but was able to significantly enhance the survival over gemcitabine and docetaxel therapy groups. In the process to delineate the mechanism for enhancement in antitumor activities of chemotherapy agents by addition of antiangiogenic agents, we demonstrate an increase in gemcitabine levels within both plasma and tumor. The observed higher fold change in gemcitabine concentration after antiangiogenic agents within tumors compared to plasma can in part be explained by a longer half-life of gemcitabine within tumor due to a greater intratumoral retention of gemcitabine after dissipation of the earlier peak levels within plasma and tumor {Dineen, 2010 #31}. Aside from this enhanced retention of gemcitabine in tumors, a functional normalization of the tumor vasculature leading to higher uptake of gemcitabine in tumors in the presence of antiangiogenic agents, will likely be another reason for this phenomenon. Although the exact mechanisms for the enhancement of cytotoxic therapy response by antiangiogenic agents remains unclear, some other possibly contributing factors may include reduced tumor vessel leakiness with a decrease in interstitial pressure, normalization of tumor microvessels, reduction in desmoplastic tumor stroma and greater efficacy in growth inhibition of other tumorpromoting cell types within the tumor microenvironment [34]. The assumption of growth inhibitory effects of antiangiogenic agents on non-epithelial cellular components of the tumor microenvironment such as endothelial cells and fibroblasts can be supported by in vitro data in the present study, and by several previous studies from our lab [26-28,32].
In any prolonged therapy approach, the risk of cumulative toxicity is not negligible and therefore the benefits of this approach might have to be balanced with the potential for possible rate-limiting metabolic side effects. In our studies we used twice a week doses of gemcitabine at 80% of the maximum tolerable dose and docetaxel at 50% of its maximum tolerable dose and did not observe any significant drugrelated toxicity. Antiangiogenic agents, bevacizumab or EMAP, were also well tolerated during therapy period.
After observing in vivo benefits of enhanced dose intensity of cytotoxic agents through prolonged therapy and/or combination with antiangiogenic agents, we attempted to recreate some in vitro conditions to study the impact of drug exposure time on different cell types related to PDAC tumor microenvironment. In PDAC epithelial cells AsPC-1, gemcitabine and docetaxel caused more inhibition in cell proliferation at longer incubation time, supporting in vivo prolonged therapy benefits. These PDAC cells are not further sensitized by exposure to antiangiogenic agents, corroborating with our previous studies [26,28,32]. This fact is also not altered by prolonged exposure, suggesting that prolonged in vivo cytotoxic therapy benefits may be derived from greater effects against epithelial tumor cells over time, and that the enhanced combination therapy benefits likely result from effects on other stromal components. Dense desmoplastic stroma surrounding malignant epithelial cells is the histological hallmark of PDAC. It contains several different cellular components including endothelial cells and fibroblasts [35]. Targeting these endothelial cells and fibroblasts within the tumor microenvironment is a sensible approach that carries the potential to enhance traditional cytotoxic approaches that are mainly directed against epithelial tumor cells. So far this strategy has shown some promising results for solid tumor treatment [36,37], although it should be stated that some antistromal in vivo models have also led to the opposite phenomenon, i.e. increased tumor progression with worsened survival outcomes [38,39]. In representative endothelial cells and fibroblasts, we observed a significant impact of gemcitabine at 72 hours of exposure time, and addition of antiangiogenic agents caused additive effects on inhibition in cell proliferation. Our relatively simplified in vitro approach to analyze combination exposure effects on various cell types cannot be used to draw definitive conclusions about combination mechanisms within tumors; however, all findings support the notion that increased cytotoxic drug availability through antiangiogenic effects, greater drug delivery through prolonged administration and combination toxicity effects on non-epithelial cell types partake in these mechanisms. The net results of such possible mechanisms obviously can create enhanced antitumor benefits that should be of interest for possible clinical treatment designs.
In summary, we report for the first time that increased bioavailability of gemcitabine or docetaxel through either prolonged therapy and/or addition of polymechanistic antiangiogenic agents caused significantly higher antitumor effects in experimental PDAC. Results observed after either approach appear to be a due to combined effects against epithelial and stromal cellular tumor components, and are sufficiently encouraging to be explored for clinical applications.
Dr. Roderich Schwarz is on the speakers’ bureau for Genentech. This does not affect the presented experiments and results.