Journal of Depression and Anxiety
Open Access
ISSN: 2167-1044
ISSN: 2167-1044
Review Article - (2014) Volume 3, Issue 1
Depression is often associated with local and/or systemic diseases. Chronic inflammation, pain and depression are a clinical triad increasingly recognized as co-morbid. Peripheral and central inflammation can alter neuronal activity, thereby contributing to the psych emotional and somatic symptoms of depression. Preliminary evidence suggests that mast cells direct the immune pathways mediating this triad. Chronic pelvic pain in women could be a paradigm of the triad, and of considerable clinical relevance. Mast cells are immune cells which contribute to and modulate inflammation and immunity. Upon stimulation mast cells release an array of mediators, cytokines, and growth factors to orchestrate an inflammatory response. These mediators can directly initiate tissue responses on resident cells, and may also regulate the activity of other immune cell functions, including central nervous system immune cells like microglia. Mast cell mediators can interact also with neurons, either alone or in concert with other immune cells. New evidence supports the involvement of peripheral and central mast cells in the development of pain processes, in the transition from acute, nociceptive to chronic and neuropathic pain and in the depressive states associated with them. A significant increase of pro-inflammatory cytokines is evident in depression associated with chronic diseases. Marked increases of tissue mast cells and pro-inflammatory cytokines have been documented in disorders associated with chronic pelvic pain presenting as well with significant affective comorbidities (anxiety and depression). Mast cells could be thus considered as a target of therapeutic approaches which aim to reduce inflammation and the chronicization of pain. The present review analyzes current evidence on co-morbidity between depression with peripheral and central inflammation associated with chronic pelvic pain in women. The analysis will briefly focus on key mechanisms of inflammation and role of mast cells as a common denominator of depression and chronic pain. The goal is to offer mental health professionals a biologicallyoriented contribution in the pathophysiologic reading of co-morbidities between depression and chronic pelvic pain in women. Results underline the increasing evidence supporting a role of inflammatory pathways/mechanisms, orchestrated by activated mast cells, as common denominators contributing to co-morbidity between depression and chronic pelvic pain in women.
Keywords: Mast cells, Chronic pelvic pain, Anxiety, Depression, Comorbid diseases, Neuropathic pain
Depression, once considered a major psychological disorder is now evolving into a more biologically-oriented pathophysiologic description [1,2]. While depression can, no doubt, be provoked by psychoemotional factors (personal, relational and/or context-dependent), the identification of the underling biologically-mediated nervous and systemic changes is important to properly manage it [3]. Depression is often associated with local or systemic diseases [4,5]. Peripheral organ inflammation and neuroinflammation can alter neuronal activity both within and outside the brain, thereby contributing to the psychoemotional and somatic symptoms of depression [2,4].
Chronic pain and underlying inflammation frequently co-occur with mood disorders such as anxiety and depression and show a high prevalence in women affected by pelvic/gynecologic pathologies [6-10]. This is probably due to the greater vulnerability of women to developing depression as compared to men. Moreover, women are more likely to report emotional distress associated with their pain [4,11]. Further, alterations in pain perception and tolerance to pain were reported in depressed patients not suffering from chronic pain [12].
Chronic inflammation, Chronic Pelvic Pain (CPP) and depression co-morbidity suggests the existence of common pathogenic mechanisms whose identification can facilitate diagnostic and therapeutic strategies for intervention. Immune system dysregulation is emerging as a common event in these conditions. In particular, Mast Cells (MCs) are now considered as cellular sensors in inflammation and immunity [13], as coordinators of peripheral inflammatory processes [14], and as important players in the development and maintenance of neuroinflammation due to their capacity to directly or indirectly interact with glial cells [15-17]. MCs might pilot the persistent lowgrade inflammation which is present both in chronic pain and mood disorders such as anxiety and depression. This evidence opens the possibility to target immune cells, particularly MCs, to act on common mechanisms of mood disorders and chronic pain. Supported by these findings, the aim of this review is to update the main evidence supporting existence of a common neuro-immune dysregulation, mainly driven by MC activation, in patients affected by depression associated with CPP.
CPP, as defined by the International Association for the Study of Pain is chronic or persistent pain, of six months duration or more, perceived in structures related to the pelvis of either men or women”. Its description includes the frequent association with negative cognitive, behavioral, and emotional consequences [18]. However, the frequency and implications of co-morbid pain and depressive symptoms among female patients in women’s health settings is relatively unknown. Moreover, the few available studies are rather heterogeneous and results are not always comparable. Poleshuck [8] reported that nearly 10% of gynecology outpatients sampled exhibited co-morbid high depressive symptoms and pain. Anxiety disorders were highly prevalent among women with co-morbid pain and depression. The co-occurrence of depression and pain doubled in economically disadvantaged patients [19]. Depression was detected in 86% of women with CPP associated with symptomatic endometriosis [20], while it was present in a significantly lower percentage (38%) in patients with asymptomatic endometriosis. In line with these observations, 86.5% and 87.5% of women diagnosed with pelvic endometriosis (80.8% of which referred pelvic pain) presented depressive symptoms and anxiety, respectively [21]. In the case of vulvodynia, current and lifetime prevalence rates for major depressive disorder were 17% and 45%, respectively. Women with current major depressive disorder reported significantly greater pain severity, and worse functioning and quality of life than women without current major depressive disorder. Among those with lifetime major depressive disorder, the majority reported their first depressive episode to have occurred before onset of vulvodynia, thus suggesting that psychiatric morbidity plays an important role in the pathogenesis of vulvodynia [22,23]. Depression or anxiety symptoms not only were more prevalent in women with vulvodynia than without, but the odds of vulvodynia were 4 times greater among women with antecedent mood disorders or anxiety [24]. The high rates of probable current depression in women with bladder pain syndrome/interstitial cystitis (BPS/IC) symptoms has been recently confirmed by a telephone screening of 146,231 households, which showed one-third of patients to have a probable diagnosis of depression [25]. BPS/IC has been confirmed as associated with anxiety disorder, even after taking demographic characteristics, medical co-morbidities, and substancerelated disorders into consideration. Anxiety disorder was found in 16.2% of women affected by BPS/IC as compared to 3.6% in controls [26]. Among diseases with CPP, current irritable bowel syndrome (IBS) in women was consistently associated with anxiety and mood disorders (27.5%) as well as lifetime IBS (50.5%) [27].
Although further studies to evaluate the prevalence of co-morbid pain and mood disorders in women with CPP are warranted, the available data confirm high prevalence rates, similar to other primarycare settings [28]. However, the findings that depression reduces pain threshold and increases pain perception, and that chronic pain facilitates the development of depression confirm that depression and chronic pain might share some common pathogenic mechanisms.
MCs behave as cellular sensors, directing tissue responses in peripheral inflammation [13,14]. Their activity can be initiated after tissue injury [13], in response to certain metabolic conditions [29], through hormonal signals [30], or by signals originating from the nervous system [31]. When activated, MCs rapidly release preformed mediators that include histamine, heparin, serotonin, chemotactic factors and various proteases such as peroxidase, tryptase, chymase, carboxidase, and beta-glucuronidase, and tumor necrosis factor alpha (TNF-α) [32]. MCs generate and release abundant quantities of newly formed mediators such as prostaglandins, leukotrienes, numerous cytokines (e.g. interleukins (IL)-1, -3, -4, -5, -6, -10, -4 and -17), and trophic factors. MCs migrate to the inflammatory site where they interact with other tissue resident or infiltrating cells [33]. By releasing cytokines and/or fostering cross-talk with other immune cells, MCs mediate immunomodulatory functions [15,34]. MCs, however, play also a primary role in the resolution of inflammation and maintenance of tissue homeostasis [35].
Dysregulation of MC function by chronic stimulation and/or defective control mechanisms can promote pathology and contribute to low grade and chronic inflammation and auto-immune diseases. MC dysregulation is frequently associated to alterations of nerve terminal fiber function and density, a condition that facilitates the onset of chronic and neuropathic pain.
Disorders associated to CPP are characterized by this neuroimmune dysregulation, involving mainly MC distribution/function and nerve terminal fibers.
Elevated numbers and activation of MCs have been consistently reported in endometriotic tissue as compared to normal or eutoptic endometrial tissues [36,37]. This MC augmentation is more evident in deep infiltrating lesions (typically associated with more severe pelvic pain) and in close proximity to nerve fibers. A concomitant alteration of somatosensory fibers, namely an augmentation of nerve fiber density, parallels MC alteration in the affected tissues [38]. BPS/IC has also been reported to show an accumulation and activation of MCs. MCs number increases in bladder, mainly in the detrusor, lamina propria, and submucosa [39,40]. Clinical studies have confirmed increased urine levels of MC mediators, including histamine [41], tryptase [42], IL (interleukin)-6 [43] and IL-8. Furthermore, serum and urine levels of YKL-40, a new biomarker expressed in bladder MCs, may serve as an index of bladder fibrogenesis as bladder “cyto-architecture” is progressively disrupted by chronic inflammatory processes. Functional urothelium and wall tissues are replaced by a connective a-functional tissue with progressive scarring until the final, irreversible stage of IC with its rigid bladder wall is reached. Activation of urinary bladderassociated circuits in the Central Nervous System (CNS) may initiate substance P (SP) release from peripheral nerves in the bladder, thereby promoting SP-mediated MC activation. MC activation may, in turn, induce bladder inflammation by acting on the urothelium. While histamine has been postulated to modulate pelvic pain, TNF-α seems to be involved in pathophysiological changes in the urinary bladder, including inflammatory changes [40,44].
MCs have been reported to play an important role in the inflammatory processes described in IBS. In this latter setting, inflammation can be triggered by food, immuno-allergic factors, infections, antibiotics, disruption of colonic ecosystems and/or systemic pathologies. Stress can further contribute through the corticotropinreleasing pathway by provoking MC degranulation. IBS patients have increased serum concentrations of IL-8, a cytokine primarily responsible for attraction of MCs and granulocytes [45]. While reports on absolute MC numbers in the intestine vary considerably, they are qualitatively consistent in documenting an increase [46-48]. Increased numbers of MCs associated with elevated numbers of serotoninergic cells have been demonstrated in colonic biopsies from IBS patients, suggesting that release of serotonin directly or indirectly from intestinal MCs may be responsible for sensory neuron activation and abdominal pain in this pathology [49]. MC tryptase and histamine may also activate enteric nerves, resulting in neuronal hyperexcitability [50,51]. In humans, degranulation of MCs in close proximity to nerves innervating the colonic mucosa correlates with abdominal pain in IBS patients. Moreover, the density of colonic tissue MCs in IBS shows a linear relationship with pain intensity [46].
In diseases associated to CPP, MC mediators can significantly influence inflammatory responses by contributing to changes in the extracellular matrix, as well as to alterations in the numbers, structure, and function of cells that are normally resident in the affected sites, including epithelial cells, vascular endothelial cells, vascular and smooth muscle cells, fibroblasts, and nerves [39,40,52,53]. The principal evidence in support of neuroimmune dysregulation in patients affected by disorders associated to CPP is summarized in Table 1.
| Pelvic disease | General features | Neuroimmune involvement | Ref. |
|---|---|---|---|
| Endometriosis | Leading etiology of CPP, estimated prevalence of 1-2% | •Elevated numbers and activation of MCs in endometriotic tissue •Stem cell factor, the major growth, differentiation and chemoattractant factor for MCs, is found in higher concentrations in the peritoneal fluid of patients affected by endometriosis •Concomitant alteration of somatosensory fibers, namely an augmentation of nerve fiber density, parallels the alteration of MCs in the affected tissues |
36-38, 52, 108 |
| Vulvar vestibulitis-provoked vestibulodynia | Most frequent etiology of coital pain located at the entrance of the vagina (“introital dyspareunia”) during the fertile age | •Tissues from subjects with localized vulvodynia display significant increase in vestibular MC number, paralleled by subepithelial heparanase activity •Dysregulation of MC activity and nerve terminal density •Subepithelial hyperinnervation and MC degranulation in localized vulvodynia, as well as in primary and secondary vestibulodynia •Augmentation of somatosensory nerve fiber density parallels increased number of MCs and degranulating MC in the affected tissue vulvar vestibulitis |
3, 109-112 |
| Bladder pain syndrome/interstitial cystitis (BPS/IC) | Mainly occurs in females and its weighted prevalence estimates for the high sensitivity and specificity definitions were 4.2% and 1.9%, respectively | •Accumulation and activation of MCs in the bladder, especially in patients with IC •Increased urine levels of MC mediators (histamine, tryptase, IL-6, IL-8) •Serum and urine levels of YKL-40, a new biomarker expressed in bladder MCs (potential index of bladder fibrogenesis) •Activation of urinary bladder-associated circuits in the CNS may initiate SP release from peripheral nerves in the bladder, thereby promoting SP-mediated MC activation and bladder inflammation •TNF-α is involved in pathophysiological changes in the urinary bladder, including those of an inflammatory nature •Activation of urinary bladder-associated circuits in the CNS may initiate SP release from bladder-derived peripheral nerves and SP-mediated MC activation |
39-44, 113-115 |
| Irritable bowel syndrome (IBS) | Inflammation can be triggered by food, immuno-allergic factors, infections, antibiotics, disruption of colonic ecosystems and/or systemic pathologies | •Stress contributes through the corticotropin-releasing pathway by provoking MC degranulation •IBS patients have increased serum concentrations of IL-8 (MC and granulocyte chemoattractant) •Increased MC numbers in the intestine of IBS patients •Increased numbers of MCs associated with elevated numbers of serotoninergic cells in colonic biopsies from IBS patients (release of serotonin directly or indirectly from intestinal MCs may be responsible for sensory neuron activation and abdominal pain) •MCs and histamine may also activate enteric nerves, resulting in neuronal hyperexcitability •In humans, degranulation of MCs in close proximity to nerves innervating the colonic mucosa correlates with abdominal pain in IBS •The density of colonic tissue MCs in IBS shows a linear relationship with pain intensity |
45-50 |
Table 1: The most frequent pathological conditions associated with CPP in women: evidence for neuroimmune dysregulation.
Collectively, data concerning the role of MCs in inflammatory processes associated to CPP suggest that they not only coordinate the initiation of inflammatory response but also have an important influence on key processes within chronically inflamed tissues, including leukocyte recruitment and activation, fibroblast proliferation, angiogenesis, matrix remodeling, and injury to affected areas.
Recent evidence supports the hypothesis that stress, anxiety and depression are associated with dysregulation of innate immune responses resulting in inflammation which, in turn, may contribute to the development of a number of disorders as well as alterations in behavior [54]. MCs might well contribute importantly to this dysregulation.
In the brain, MCs reside on the brain side of the blood-brain barrier and interact with neurons, glia, blood vessels, and other hematopoietic cells via their neuroactive prestored and newly synthesized mediators [13,14]. They are first responders, acting as catalysts and recruiters to initiate, amplify, and prolong other immune and nervous responses upon activation [16]. MCs might therefore represent the immune cells that peripherally and centrally coordinate inflammatory processes in neuropsychiatric diseases. Brain MCs respond to immune and nonimmune signals such as Corticotropin Releasing Hormone (CRH), various neuropeptides and neurotensin. In particular, CRH secreted under stress can stimulate MCs through a synergistic action with neurotensin to increase vascular permeability and disrupt the bloodbrain barrier [30]. Acute stressors or CNS injury can change both phenotype and numbers of brain MCs [55,56]. Activation of brain MCs releases neuroactive mediators into the brain parenchyma which may be linked to emotionality. Indeed, patients with MC-mediated diseases such as food allergies, asthma, and IBS often complain of associated anxiety [57,58]. Moreover, patients with systemic mastocytosis also report low arousal states, lethargy, and coma [59,60].
MC-dependent effects on behavior may be mediated by multiple interacting messengers and neural systems. For example, histamine has both anxiolytic and anxiogenic effects, with opposing roles attributed to H1 versus H2 receptors [61,62]. Serotonin functions both as a transmitter affecting aggression, appetite, and mood, and as a trophic factor influencing neurogenesis to affect emotionality and memory [62]. Selective serotonin reuptake inhibitors increase serotonin signaling and decrease anxiety; therefore, a lack of MC-derived serotonin may increase anxiety-like behaviors [63]. MC-derived cytokines act as neuromodulators, affecting systems controlling behavior. Indeed, TNF-α, IL-1, and IL-6 act on the hypothalamic-pituitary-adrenal axis to control stress behavior. In addition, MCs express receptors for, and can be stimulated by CRH, with the release of histamine, IL-8, tryptase and vascular endothelial growth factor [64]. Given their repertoire of mediators, it would not be surprising for MCs to have multifaceted interactions with brain systems controlling behavior.
The ability of peripheral MCs to coordinate and drive inflammatory processes allows these cells to profoundly affect nociception and pain signaling, while CNS-located MCs have been suggested to participate in the central integration of pain. In physiological conditions, pain information begins at nociceptors, which form a functional pain unit with the nearby tissue capillaries. Peripheral MCs are important players of this functional unit, as deduced from their strategic position in proximity to sensory nerve endings and vasculature [65]. In this context MCs can be thought of as immunocytes with secretory functions that act locally in peripheral tissues to modulate local hemodynamics, nociceptor activation and pain. Following injury or inflammatory stimuli, MC mediators such as bradykinin, prostaglandins and histamine are released and stimulate nociceptive afferents. Neuropeptides such as SP may also cause MC degranulation, creating a bidirectional positive feedback-loop [66] to up-regulate local inflammation and increase pain. MC recruitment of other immune cells, which release pro-nociceptive mediators, can affect not only injured zones but also adjacent territories, creating a secondary, widespread hyperalgesia [67]. The contribution of meningeal MCs to the activation of meningeal nociceptors in the pathophysiology of migraine has been extensively documented [31,68]. At the spinal level, mediators released from dural MCs may reach the superficial laminae to modulate synaptic transmission and nociception [69]. Brain MCs are particularly concentrated in the thalamus, an essential nociceptive relay where, by releasing mediators such as histamine or serotonin they might interact with third-order neurons targeting the cortex [17]. Increased numbers of MCs are found in the contralateral thalamus after nerve ligation, paralleled by development of mechanical hyperalgesia [70]. In addition, cerebral MCs may play a role in nociception: spinal application of supernatant from activated cultured MCs reportedly induced significant mechanical hyperalgesia, long-term potentiation at the spinal synapses of C-fibers and increased numbers of MCs in the lumbar, thoracic and thalamic areas [71]. Interestingly, nociceptive signals influence the distribution of MCs in the brain during development of hyperalgesia: MCs infiltrate the thalamus from blood to brain within a few hours in response to chemotactic molecules directly released by peripheral sensory afferents [72]. Finally, the infiltration and degranulation of thalamic MCs is reportedly influenced by molecules known to act on nociception such as sumatriptan, whose administration doubles the number of degranulated MCs [73].
Besides MC-mediator activation of neurons, cell-to-cell communication between MCs and neurons can operate via adhesion molecules expressed on their surfaces, such as cell adhesion molecule-1 and N-cadherin [31,51,74]. Moreover, bidirectional crosstalk between MCs and microglia (the brain’s major resident immune cell) has been reported [15], proposing that MCs might act in synergy with glial cells to influence central elaboration of pain signaling [75]. MC involvement in development and progression of pain in disorders associated to CPP has been demonstrated. MC degranulation at the visceral level increases excitability of vagal, splanchnic and mesenteric afferents, contributing to nociceptive processes associated with visceral pain. Of note, women experiencing CPP often exhibit overlapping symptoms of BPS/IC and IBS [76]. Much of this overlap can be attributed to central and peripheral neural mechanisms of pelvic organ cross-sensitization [77-79]. However, the juxtaposition of SP-positive nerve fibers and MCs [80] may play an important role in pelvic organ cross-sensitization by amplifying afferent signaling. In fact, MC pro-inflammatory mediators can affect not only injured zones, but also adjacent territories, creating a secondary, widespread hyperalgesia [81]. The contribution of MCs to colon–bladder cross organ-sensitization induced by application of a colon chemical irritant has been confirmed using the MC stabilizing agent and histamine H1 agonist ketotifen fumarate [82].
The co-occurrence of CPP and depression suggest one or more of common mechanisms underlying their co-morbidity. Biological mechanisms behind both pathologies include neurotransmitter reduction (mainly serotonin and norepinephrine), imbalance in peripheral and central cytokines and Hypothalamic-Pituitary-Adrenal Axis (HPA) dysfunction. Importantly, MCs are involved in all of these mechanisms (Figure 1).
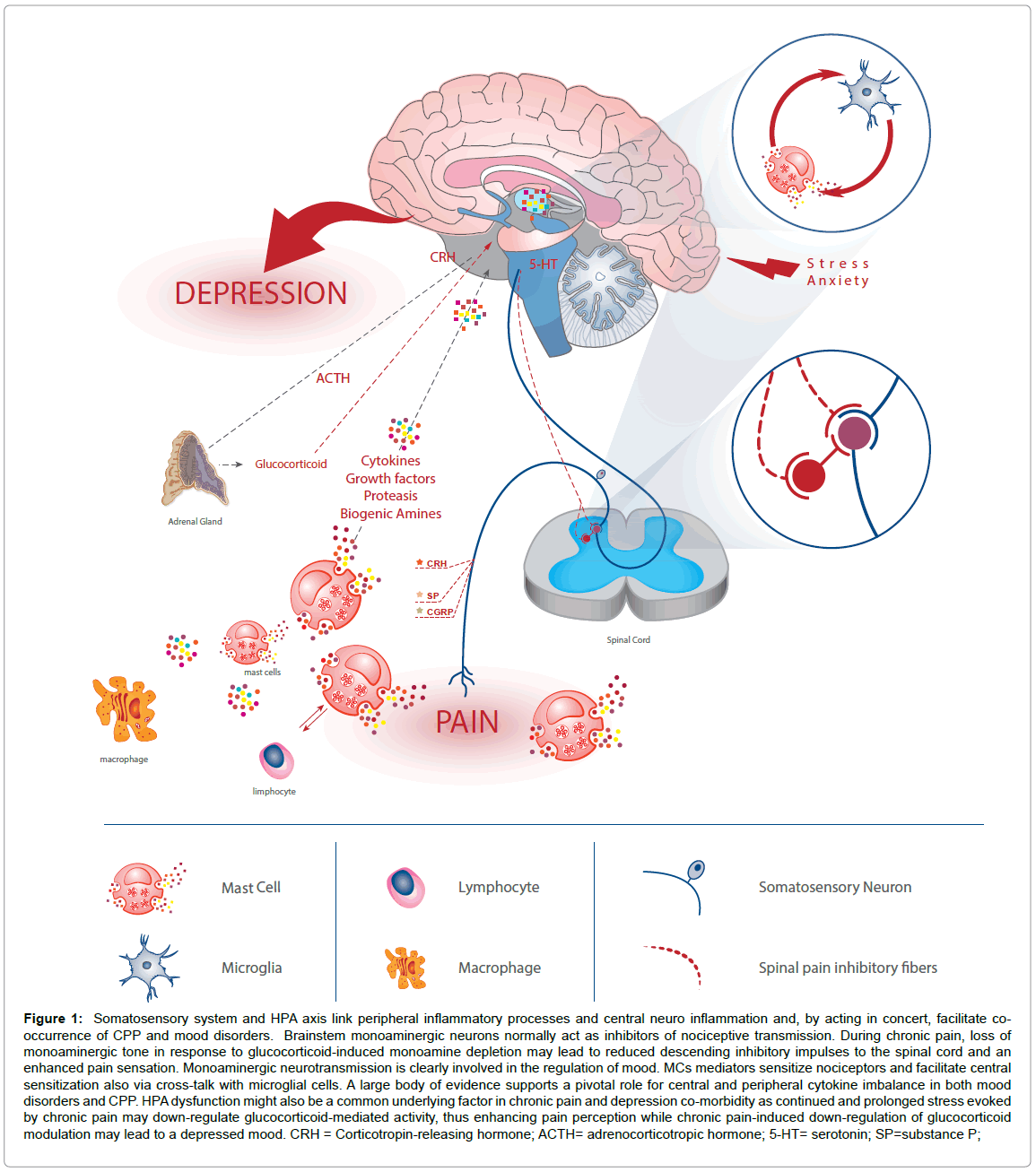
Figure 1: Somatosensory system and HPA axis link peripheral inflammatory processes and central neuro inflammation and, by acting in concert, facilitate cooccurrence of CPP and mood disorders. Brainstem monoaminergic neurons normally act as inhibitors of nociceptive transmission. During chronic pain, loss of monoaminergic tone in response to glucocorticoid-induced monoamine depletion may lead to reduced descending inhibitory impulses to the spinal cord and an enhanced pain sensation. Monoaminergic neurotransmission is clearly involved in the regulation of mood. MCs mediators sensitize nociceptors and facilitate central sensitization also via cross-talk with microglial cells. A large body of evidence supports a pivotal role for central and peripheral cytokine imbalance in both mood disorders and CPP. HPA dysfunction might also be a common underlying factor in chronic pain and depression co-morbidity as continued and prolonged stress evoked by chronic pain may down-regulate glucocorticoid-mediated activity, thus enhancing pain perception while chronic pain-induced down-regulation of glucocorticoid modulation may lead to a depressed mood. CRH = Corticotropin-releasing hormone; ACTH= adrenocorticotropic hormone; 5-HT= serotonin; SP=substance P;
Brainstem monoaminergic neurons normally descend to the spinal cord to act as inhibitors of nociceptive transmission. During chronic pain, loss of monoaminergic tone in response to glucocorticoidinduced monoamine depletion may lead to reduced descending inhibitory impulses to the spinal cord and an enhanced pain sensation (Figure 1). Monoaminergic neurotransmission involvement in the regulation of mood as a reaction to psychological stress has been long known. Since serotonin reuptake inhibitors, by increasing serotonin signaling, decrease anxiety, it is likely that a lack of MC-derived serotonin might increase anxiety-like behaviors. Cytokine imbalance caused by dysregulation of immune cells, particularly by MCs (see before) orchestrates inflammatory responses. The ensuing increase in pro-inflammatory agents and nerve growth factor amplifies pain processes via a sensitization of somatosensory neurons [75]. A large body of evidence supports a pivotal role for central and peripheral cytokine imbalance in both mood disorders and CPP (Figure 1). HPA dysfunction might also be a common underlying factor in chronic pain and depression co-morbidity: continued and prolonged stress evoked by chronic pain may down-regulate glucocorticoid-mediated activity, thus enhancing pain perception. Similarly, chronic pain-induced down-regulation of glucocorticoid modulation may lead to a depressed mood. Women with CPP and low depression exhibit enhanced adrenocorticotropic hormone responses to psychosocial stress compared with those having CPP and high depression, whereas there were no differences in cortisol responses. In the adrenocorticotropic hormone1–24 stimulation test, CPP patients with high depression demonstrated enhanced cortisol responses [83]. These findings support a relationship between depression and HPA reactivity in patients with CPP. It thus appears that the somatosensory system and HPA axis, and their interaction with the immune system, may form a cycle connecting peripheral inflammatory processes and central neuro inflammation that facilitate co-occurrence of CPP and mood disorders.
Depression has a solid inflammatory basis. MCs appear to trigger and modulate the general inflammatory process in every tissue. The pelvis is no exception, where MCs direct the shift towards chronic and neuropathic pain. This shift is more likely when the etiology of inflammation remains undiagnosed/neglected/undertreated, and/or when patients are genetically disposed towards hyperactive immune-allergic responses. The cornerstone of an etiologicallyoriented treatment would be to act on predisposing, precipitating and maintaining factors which contribute to chronic pain, an exemplified in vulvodynia patients [3]. MCs are, in essence, the biological correlate for each of these factors. As such their modulation represents a critical target for reducing the cascade of events that, if left untreated, can lead to chronicization of inflammation and pain. Indeed, a number of studies demonstrate that pharmacologically blocking MC activation reduces pelvic inflammation and pain. For example, oral administration of ketotifen normalized bladder capacity and permeability in trinitrobenzene sulfonic acid-treated rats, thus reducing colon–bladder cross-organ sensitization [82]. The peripherally restricted secondgeneration H1 receptor antagonists fexofenadine and ebastine reversed post-stress visceral hypersensitivity in rats [84]. Moreover, systemic treatment with the MC stabilizer sodium cromoglycate attenuated restraint stress-associated behavioral alterations [85]. The underlying mechanisms of these effects remain unclear, as treatment did not alter MC numbers or their secretion of histamine or tryptase [86].
Among agents active on MCs, Palmitoylethanolamide (PEA) is of particular interest. PEA, an endogenous fatty acid amide, is a congener of the endocannabinoid anandamide belonging to a class of lipid mediators, the family of N-acylethanolamines. PEA has been reported to reduce pain behaviors, and to inhibit somatosensory activation and correlated events in different models of pelvic inflammation [87-97]. Moreover, PEA exerted an antidepressant-like effect comparable to the reference drug fluoxetine [98,99] (Table 2). In Italy and other European states, products based on PEA are approved as therapeutics classified as “dietary foods for special medical purposes”. The combination of micronized PEA and polydatin, in particular, provides significant and long-lasting relief of pelvic pain symptomatology (Table 3) [100-105]. Given the optimal safety profile of PEA, products based on this fatty acid amide may find utility as tools to control pain in pelvic disorders associated to CPP, as well as co-morbid anxiety and depression.
| Model | PEA: administration route and dosage | biological action | Ref. | |
|---|---|---|---|---|
| Bladder | turpentine instillation into the urinary bladder | 10-30 mg/kg ip | •attenuates the viscero-visceral hyper-reflexia | 87 |
| 10-30 mg/kg ip | •reduces thermal hyperalgesia | 88 | ||
| NGF instillation into the urinary bladder | 1.5-2.5 mg/kg ip | •attenuates bladder hyper-reflexia •reduces Fos expression in spinal cord |
89 | |
| i.pl. NGF | 10-25 mg/kg i.pl. | •decreases thermal hyperalgesia •reduces neutrophil accumulation |
90 | |
| acetic acid, kaolin and magnesium sulfate-evoked writhing | 1-20 mg/kg ip | •reduces writhing | 91 | |
| magnesium sulfate-evoked writhing | 1-100 mg/kg s.c. | •reduces writhing | 92 | |
| phenyl-p-quinone induced visceral pain | 2-8 mg/kg ip | •inhibits stretching movements | 93 | |
| chronic intestinal inflammation induced by croton oil | PEA, 2.5 – 30 mg/kg ip | •inhibition of intestinal motility | 94 | |
| Controls | 1–20 mg/kg ip | •inhibition of small intestine motility | 95 | |
| intestinal injury and inflammation caused by ischemia-reperfusion | 10 mg/kg ip | •reduction of pro-inflammatory cytokine production •reduction of histological alterations |
96 | |
| dextran sodium sulphate-induced colitis | 2-10 mg/kg ip | •improves all macroscopic signs of ulcerative colitis •decreases the expression and release of all the pro-inflammatory markers |
97 | |
| Anxiety/ depression |
Controls | 5-40 mg/kg | •reduction of immobility (in TST and FST tests | 98 |
| chronic corticosterone administration | PEA 1-10 mg/kg vs. co-ultramicronized PEA+luteolin (1 mg/kg, ip) | •attenuation of depressive-like state •increases hippocampal neurogenesis and dendritic spine density |
99 |
PEA=Palmitoyl Ethanol Amide; IP=Intraplantar; IP= Intraperitoneal; SC=Subcutaneous; NGF=Nerve Growth Factor; TST=Tail Suspension Test; FST=Forced Swimming Test.
Table 2: Principal results concerning the effects of PEA on pain behaviors, inhibition of somatosensory activation and correlated events and on anxiety-depression status.
| Source of pain | Study Design | PEA treatment | PEA effects | Ref. |
|---|---|---|---|---|
| Endometriosis | Open (case series) micronized PEA | micronized PEA 200 mg+Polydatin 20 mg/bid for 3 months | •decreased pelvic pains: •CPP •dyspareunia •dysmenorrhea •decreased use of analgesics. |
100 |
| Endometriosis | Double blind, randomized parallel-group, micronized PEA vs placebo | micronized PEA 400mg+40mg/ Polydatin/tid) for 3 months vs. celecoxib 200 mg/bid for 7 days | •decreased pelvic pains: •CPP •dyspareunia •dysmenorrhea |
102 |
| Endometriosis: •recto-vaginal septum(19) • ovary(28) |
Prospective micronized PEA | micronized PEA 400mg+40mg/ Polydatin//bid) for 3 months | •decreased pelvic pains: •CPP •dyspareunia •dysmenorrheal •dyschezia |
103 |
| Endometriosis | Prospective micronized PEA | micronized PEA 400 mg+40 mg/ Polydatin//bid) for 3 months | •decreased pelvic pains: •CPP •dyspareunia |
104 |
| Pudendal neuralgia | Case Report micronized PEA | 300 mg micronized PEA/tid gradually decreasing to 300 mg/die for1 year | resolution of pain | 101 |
| Vestibulodynia | Double blind randomized parallel-group, TENS vs. TENS + micronized PEA | 400 mg micronized PEA+Polydatin 40 mg/bid | Improvement of pain relief | 105 |
CPP= Chronic Pelvic Pain; PEA=Palmitoyl Ethanol Amide; TENS=Transcutaneous; Bid= Twice Daily; Tid= Three Times a Day
Table 3: Clinical studies showing analgesic effect of micronized PEA in conditions associated to chronic pelvic pain
A female patient complaining of pain, and specifically of CPP, should no longer be told that “pain is all in your head”, “pain is psychogenic”, “you are inventing your pain” and similar other unfounded statements [3], as they ignore the biological substrates of pain. Moreover, such statements reduce the therapeutic potential of a correct biological reading of depression and pain by banalizing both.
In parallel, the increasing evidence that an appropriate psychotherapeutic regimen can reduce inflammation, depression and pain [106-115] should encourage a more synergistic treatment approach, by “giving words to pain” to relieve the emotionally intimate burden and its biological correlates, while modulating biological common denominators of the triad (inflammation, depression and CPP) with therapeutic approaches which target a reduction in MC hyperactivity. The final goal is to empower and accelerate the healing process of depression and associated co-morbidities with a womancentered, comprehensive and biologically-based approach.
This study was supported in part by MIUR, PON ‘Ricerca e Competitivita` 2007–2013’ project PON01_02512.