Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
![]() +44 1300 500008
+44 1300 500008
ISSN: 2157-7064
![]() +44 1300 500008
+44 1300 500008
Research Article - (2015) Volume 6, Issue 7
The issue of selectivity due to ion suppression or enhancement caused by the sample matrix has become a challenge in mass spectrometric (MS) analysis. It has been pragmatic that an efficient sample preparation method can reduce the matrix effect significantly. The study was planned to assess the effect of ionization type and sample preparation techniques on the presence or absence of matrix effect in quantitative liquid chromatography-tandem mass spectrometric (LC-MS/MS) analysis of bosentan (BSN) by post extraction addition method. Different sample processing techniques, i.e., protein precipitation (PPT), liquid–liquid extraction (LLE), and solid phase extraction (SPE) were evaluated. The sample were analyzed by both LC-electrospray ionization (ESI)-MS/MS and LCatmospheric pressure chemical ionization (APCI)-MS/MS. Chromatographic partition achieved on an Aquasil C18 column (100 × 2.1 mm, five μm). The mobile phase consisted of ammonium formate (pH 4.5) and methanol (10:90 v/v) in non-gradient elution mode. Our results demonstrated that both ionization sources showed matrix effect, but ESI was more prone to matrix effect than APCI. Sample processing techniques could decrease or increase matrix effect. PPT was found to be least efficient sample preparation technique, due to the presence of many remaining matrix constituents and often resulting in significant matrix effects. LLE also provided clean final extracts. However, the efficiency of LLE was lower than SPE. SPE methods resulted in cleaner extracts and considerably reduced the levels of residual matrix components from plasma samples that ultimately leads to significant reduction in matrix effects. LC-APCI-MS/MS was found to be the ionization of choice for quantitative analysis of BSN and other drugs with similar physicochemical properties. The combination of SPE and the ionization source offer a major advantage in reducing matrix effects resulting from plasma components and in improving the selectivity and sensitivity of drug analysis.
Keywords: Bosentan; Matrix effect; Ionization source; Plasma extraction; Mass spectrometry
LC-MS-MS is a powerful analytical tool for quantitative drug analysis due to its high sensitivity, selectivity, and specificity. It is allowing for the analysis of trace amounts of intended analytes in complex mixtures [1-3]. By these characteristics, one can expect simplification of sample preparation and less time-consuming method and sample analysis time during routine analysis. However, the limitation associated with LCMS analysis is its susceptibility to a matrix effect [4]. Matrix effect is the impact of eluting remaining matrix components on the ionization of the compound of interest [5,6]. Typically, diminished precision and accuracy of subsequent measurements are the results of suppression or enhancement of analyte response [5]. Matrix effect caused by several factors, including endogenous phospholipids, dosing media, formulation agents, and mobile phase modifiers [7]. Matrix effects cause either in ion suppression, or, in some cases, ion enhancement. It can be highly unpredictable and can be difficult to control or foresee [8].
The magnitude and nature of suppression or enhancement are the functions of the concentration of the co-eluting matrix constituents. Moreover, matrix effects are analyte specific, and several ways have been tried to minimize/eliminate the matrix effect [9]. These include changes in sample preparation techniques, chromatographic conditions, selection of ionization source, changes in polarity, and stable isotopelabeled internal standard and decreasing the amount of sample for extraction [10]. The use of inefficient extraction process, improper ion polarity, and ionization source are the major contributors to the matrix effect [11]. However, the data on this important and synergistic effect of sample preparation and ionization source on matrix effect is limited [12]. Dams et al. determined the matrix effect for morphine by postcolumn infusion method where they continuously injected analyte after infusing an extract from a sample into the system. However, the drawback of this technique is that it does not provide a quantitative perceptive of the matrix effect observed by exact analytes [4].
Here in this study, we tried to provide a complete evaluation of both sample preparation methods and ion source optimization for reducing or eliminating matrix effects by post extraction addition method in rat plasma. In this study, we evaluated the three most widely used sample preparation techniques, i.e., SPE, PPT, and LLE. The observations obtained from this study will be used to pair the optimal sample preparation technique and ionization type for the quantitation of BSN (Figure 1) and other drugs with similar physicochemical properties.

Figure 1: Chemical structure of bosentan (CAS ID: 147536-97-8).
Materials
BSN and internal standard (IS) azithromycin obtained from Clearsynth Ltd. Mumbai India. HPLC-MS grade acetonitrile and methanol (purity 99.9%) purchased from Sigma-Aldrich, Germany. Ethyl acetate was procured from Merck Specialties Pvt. Ltd. MS grade ammonium acetate and ammonium formate obtained from Fluka Analytical, Sigma-Aldrich, The Netherlands. Formic acid (purity >98%) procured from Fluka Analytical, Sigma-Aldrich, Germany. Water used in the analysis was prepared in-house with Milli-Q water purification system procured from Millipore (Millipore Corporation, USA). Other chemicals and materials used in the study were of ACS grade from commercial sources.
Liquid chromatography conditions
Chromatographic partition was achieved on an Aquasil C18 column (100 × 2.1 mm, 5 μm particle size), attached to UHPLC of Thermo Scientific-Dionex Ultimate 3000 (Serial # 7248679, Part # 5035.9200). The UHPLC was equipped with a quaternary solvent system, an autosampler, solvent manager (Serial # 8074857, Part # 5082.0010). The mobile phase consisted of ammonium formate (pH 4.5) and methanol (10:90 v/v) in non-gradient elution mode, which was degassed. The flow rate of mobile phase was kept at 0.3 ml/min. A fixed amount of 10 μL of sample solution injected into the MS detector in each run. The total chromatographic run time was found to be 5 min. The column maintained at 35 ± 5°C, and the pressure of the system was 70 bar.
Mass spectrometry conditions
Mass spectrometric measurements were performed on Thermo Scientific, LCQ Fleet, Ion Trap mass spectrometer (Serial# LCF 10356, San Jose, CA, USA). The accurate mass and composition for the parent ions and the daughter ions were calculated using the Xcalibur software. Ionization of analytes of interest was done using electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). All MS/ MS data for BSN and IS were collected in positive ion polarity mode by selected reaction monitoring (SRM). The optimized parameter for ESI were: spray voltage 5.0 kV, sheath gas 40, auxiliary gas 5, sweep gas 0 (highly pure nitrogen). The APCI parameter settings were: vaporizer temperature 300°C, corona discharge needle voltage 5 kV, sheath gas (high-purity nitrogen) 50, and no use of auxiliary gas. Transfer capillary temperature was set at 270°C for both ESI and APCI respectively.
Sample processing
Stock solutions of BSN and IS prepared in methanol (1 mg/ ml). Working solutions of BSN was prepared at eight different concentrations (10.04-2002.42 ng/mL) in methanol–water (50:50; v/v). These samples were spiked in plasma (2% v/v) to yield calibration standards. QC samples prepared at four concentrations (10.04, 25.37, 907.10, and 1645.64 ng/mL) levels. We investigated the following three different sample processing techniques.
Liquid–Liquid extraction
For sample preparation, 200 μL of plasma was mixed with 50 μL of IS working solution (1000.0 ng/mL). To this solution, 200 μL of 5% orthophosphoric acid and 200 μL of milli-Q water was added and vortexed. This mixture was extracted with 4 mL of t-butyl methyl ether, by placing the tubes on reciprocating shaker for 20 min at 100 rpm. These samples were subjected to flash freezing (liquid nitrogen), and 3 mL of clear supernatant was separated and then dried at 45°C under a stream of nitrogen at 25 psi.
Protein precipitation
For sample preparation, 200 μL of plasma was mixed with 50 μL of IS working solution (1000.0 ng/mL). To this solution, 200 μL of 5% orthophosphoric acid and 200 μL of milli-Q water was added and vortexed. Acetonitrile (1 mL) was transferred to this mixture for protein precipitation and then vortexed and centrifuged for 5 min at 4000 rpm. The clear supernatant was separated and then dried at 45°C under a stream of nitrogen at 25 psi.
Solid phase extraction
For sample preparation, 200 μL of plasma was mixed with 50 μL of IS working solution (1000.0 ng/mL). To this solution, 200 μL of 5% orthophosphoric acid and 200 μL of milli-Q water was added and vortexed. The diluted samples were transferred to Agilent Bond Elute Plexa® cartridges (30 mg/cc) which were preconditioned with 1 ml methanol and followed by 1 ml of water. The cartridges containing the samples were centrifuged for 2 minutes at 1500 rpm. Subsequently, the cartridges were washed twice with 1 mL of water and eluted twice with 1 mL of methanol. The eluate was dried at 45°C under a stream of nitrogen at 25 psi.
Finally, the every dried residue obtained by employing above stated three methodologies was reconstituted with 500 μL of the mobile phase. Ten μL of the reconstituted sample was injected onto the column for analysis.
Matrix effect
Post-extraction addition technique was used to evaluate matrix effect. Plasma samples were prepared by reconstituting the extracted blank plasma samples with reference solution containing analyte and IS. Since extraction protocol involved terminal drying step, biological at concentrations representing the quality control (QC) concentration at low (LQC), medium (MQC), and high (HQC) levels. The neat sample was reference solution prepared at an appropriate concentration in reconstitution solution (at LQC, MQC, and HQC level) [8,12,13].
Matrix effect was calculated as per the following equation
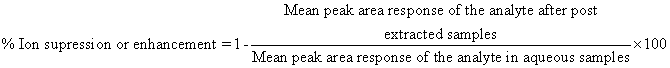
The negative value stands for the % of ion enhancement, and the positive value stand for the % of ion suppression.
Method efficiency (ME)
ME was determined by measuring the mean peak area response of six replicates of extracted QC samples (at LQC, MQC, and HQC level) against the average peak area response of aqueous solutions. ME of BSN was estimated by using the following equation:

Variables control
All the variables in the processing of samples were strictly controlled to avoid any discrepancies in the comparison of outcomes. The volume of the plasma used for the each measurement and composition of the reconstituted solvents was kept constant for all sample preparation techniques. Other variables taken into consideration were pretreatment of the samples before extraction, the volume of the reconstituted solvent, injection volume, and analytical column. Control of dilution and reconstituting solvents and their volume is necessary to keep the lipid concentrations consistent across all the sample processing techniques [1,6,14]. Pretreatment of the samples was kept uniform across processing methods to avoid any impact on final composition. The single analytical column was used throughout the analysis to achieve a uniform chromatographic separation for all the techniques. The volume of injection was constant throughout the process to ensure the same amount of sample loaded onto the column. The composition of rinsing solution and mobile phase were also kept identical throughout the analysis.
Optimizing mass spectrometric parameters
Negative matrix effect, signal suppression, and elevated background in MS analysis are due to the less purity of biological samples. To overcome these conditions the only possibility is to increase S/N and simultaneously maintaining high throughput performance by SRM mode. In the study the matrix effect was observed with ESI than APCI, i.e., with both ionization types, but was more ubiquitous with the ESI [15-17]. The ESI type of ionization was found to be highly susceptible to matrix effect due to its wide polarity range. In the presence of hydrophobic interference, the APCI was found to be less sensitive to the matrix effect. In APCI source, the BSN and IS formed positive ions under acidic conditions, induced by the mobile phase due to the addition of a proton [4,12]. Several daughter ions were observed for both, i.e., analyte as well of internal standard in the daughter ion spectra. The data for BSN and IS were collected in positive ion polarity mode at a transition of m/z 552.3-202.0 and 749.6-591.6 respectively, as these ions were copious, selective and produced unwavering responses.
Optimizing liquid chromatographic parameters
Since the study was about to compare the matrix with different conditions, it was necessary to set LC conditions to get sharp peak shape and adequate response. The selection of mobile phase buffer pH, flow rate, column type and the injection volume was utmost important. Different ratios of ACN-water and methanol-water combination tried as the mobile phase. The buffer tried were ammonium acetate, ammonium trifluoroacetate, and ammonium formate in varying strength. The final mobile phase consisted of ammonium formate (pH 4.5) and methanol (10:90 v/v) in non-gradient elution mode was found to be most appropriate for faster elution, better efficiency and peak shape (Figure 2). Aquasil C18 column (100 × 2.1 mm, 5 μm particle size) was useful in the separation and elution of both compounds in a short time (5 min.).

Figure 2: Representative chromatogram of bosentan (a) and internal standard (b) at LQC level after extraction through SPE technique and analyzed on LC-APCI-MS-MS.
Optimizing sample preparation techniques
The growing focus on sample analysis has shown the way to the regular practice of preparing samples by the simplest, fastest way possible. All the three previous stated extraction methodologies were optimized to avoid matrix effect and to get high ME across all the QC levels. During optimization, plasma was processed with different aliquot volumes to attain desired S/N ratio at LOQ level. Finally, 200 μL aliquot volumes were selected for processing through various extraction techniques.
Liquid–Liquid extraction
LLE was included in extraction techniques as it is an efficient means of sample preparation and is a cleaner option. We evaluated the reconstituted LLE extracts for matrix effects and analyte recovery [8,18,19]. As per the observations shown in Table 1, an extracting solvent for LLE process was selected based on higher % ME attained for an extracting solvent compared to the other solvents and/or solvent mixture. Thus, as per the observations the tertiary butyl methyl ether was selected as an extracting solvent. The data for optimization of pretreatment solution and volume are presented in Table 2. The pretreatment solution, i.e., 200 μL of 5% orthophosphoric acid and 200 μL of Milli-Q water was found to be suitable to attain higher % PE. However, the LLE is less realistic for many reasons. Sometimes the final extraction solvent is not compatible with initial reversed-phase LC mobile phase ratios and requires gradual removal of the supernatant [20,21].
| Extraction Solvent | (%) Recovery |
|---|---|
| Tertiary butyl methyl ether | 55 |
| Ethyl Acetate | 43 |
| Diethyl ether | 30 |
| Dichloromethane/n-hexane (50:50,v/v) | 33 |
| Ethyl acetate/N-hexane (50:50,v/v) | 39 |
Table 1: Trial for selection of solvent during LLE.
| Extraction condition | ESI | APCI | ||
|---|---|---|---|---|
| %RE | %Ion suppression | %RE | %Ion suppression | |
| 100 µL of 2% OPA and 100µL of Milli-Q water | 43 | 39 | 53 | 27 |
| 100 µL of 5% OPA and 100µL of Milli-Q water | 69 | 21 | 74 | 19 |
| 200 µL of 5% OPA and 200µL of Milli-Q water | 73 | 17.5 | 76 | 11.23 |
Table 2: Effect of extraction condition of LLE on ion suppression and method efficiency.
Protein precipitation
The PPT is the common and fastest method possible for sample extraction from a biological fluid. During PPT extraction, there was a massive reduction in %ME and enhancement of ion suppression with acetonitrile. Acidified acetonitrile with 1% formic acid resulted in substantial enhancement in the ion suppression. However, pretreatment of plasma with 200 μL of 5% orthophosphoric acid and 200 μL of milli-Q water led to decrease in matrix effect and improved ME Table 3. Although the PPT procedure is quick and easy, it does not produce an immaculate final extract. This procedure fails to remove enough of the plasma components, specifically phospholipids, which are known to cause inconsistency in analyte signal intensity in MS. The particular organic solvent used in PPT procedures has a remarkable effect on the overall cleanliness of the final extract [5,22].
| Extraction condition | ESI | APCI | ||
|---|---|---|---|---|
| %RE | %Ion suppression | %RE | %Ion suppression | |
| Acetonitrile | 46 | 41 | 52 | 36 |
| 1% formic acid in acetonitrile | 42 | 52 | 48 | 47 |
| Acetonitrile/200µL 5%OPA/200 Milli Q Water | 67 | 28 | 78 | 19 |
Table 3: Effect of extraction condition of PPT on ion suppression and method efficiency.
Solid phase extraction
For SPE, we selected an Agilent Bond Elute Plexa® (30 mg/cc) polymeric sorbent. Conventional procedure for conditioning of sorbent with methanol followed by water was carried out. The optimization of washing and elution steps was performed, which are presented in Table 4. As it is earlier reported that SPE offers cleaner extracts than PPT and LLE. Our results also demonstrated that matrix cleanup was found to be good with SPE, and pre-concentration step involved in the process did not affect the ME [23]. However, omitting a pre-concentration step results in loss of sensitivity [11,24,25].
| Washing optimization | Elution optimization | ESI | APCI | ||
|---|---|---|---|---|---|
| %RE | %Ion suppression | %RE | %Ion suppression | ||
| with 1mL water | with 1mL methanol | 83 | 8.9 | 85 | 4.2 |
| with 1mL water | with 1mL acetonitrile | 72 | 18 | 75 | 15.2 |
| with 1mL water twice | with 1mL methanol twice | 86 | 3.4 | 89 | 2.6 |
| with 1mL 2% methanol in water twice | with 1mL methanol twice | 69 | 16 | 73 | 13 |
| with 1mL 2% methanol in water twice | with 1mL2% ammoniated methanol twice | 63 | 17.6 | 57 | 19 |
Table 4: Effect of extraction condition of SPE on ion suppression and method efficiency.
Matrix effect and method efficiency
Post-extraction addition method was used to assess the ionization suppression or enhancement of the analyte at three QC concentration levels. The results in detail are presented in Table 5. Among all three techniques, minimum matrix effect was observed in SPE technique when analyzed through APCI or ESI ion source. The SPE technique combines with APCI source was found to be the best combination wherein to avoid maximum possible ion suppression. In each technique and on each ion source the matrix effect was consistent across all QC levels.
| Matrix effect/Ion suppression (%) | ||||
|---|---|---|---|---|
| Ion Source | Extraction method | QC samples | ||
| LQC | MQC | HQC | ||
| ESI | Solid Phase Extraction | 7.8 | 6.7 | 9.4 |
| Liquid Liquid Extraction | 17.1 | 18.6 | 24.4 | |
| Protein Precipitation | 22.5 | 19.2 | 28.0 | |
| APCI | Solid Phase Extraction | 4.6 | 5.2 | 5.9 |
| Liquid Liquid Extraction | 14.1 | 16.8 | 20.2 | |
| Protein Precipitation | 20.8 | 17.4 | 22.0 | |
Table 5: Ion suppression of bosentan across all QC levels after extraction with different techniques and analyzed on different ion source in MS.
The hemolysed and lipemic plasma did not cause many variations at LOQQC and HQC levels. The data for the different type of plasma are presented in Figures 3 and 4. It can be observed that SPE and LLE samples having minor variation in precision and accuracy of QC samples in comparison to PPT samples. Ionization sources the APCI was found to be having least matrix effect on the target analyte.

Figure 3: Matrix effect at LOQQC levels from different type of plasma samples extracted by different techniques (n=10).

Figure 4: Matrix effect at HQC levels from different type of plasma samples extracted by different techniques (n=10).
In all the processing techniques, maximum ion suppression was noticed in PPT, which was not affected by the type of ionization technique (APCI or ESI). In PPT, the ion suppression was mainly due to co-eluting substances in the samples during extraction that ultimately results in decreased ME in comparison to other processing techniques. The detailed observation for ME is given in Table 6. The LLE and SPE techniques were found to be having least ion suppression and higher ME, however as per the ME the best technique was found top be SPE.
| Method efficiency (%) | ||||
|---|---|---|---|---|
| Ion Source | Extraction method | QC samples | ||
| LQC | MQC | HQC | ||
| ESI | Solid Phase Extraction | 80.9 | 82.2 | 79.4 |
| Liquid Liquid Extraction | 66.8 | 56.8 | 63.0 | |
| Protein Precipitation | 15.2 | 10.2 | 28.0 | |
| APCI | Solid Phase Extraction | 84.4 | 88.7 | 83.2 |
| Liquid Liquid Extraction | 65.9 | 54.1 | 62.7 | |
| Protein Precipitation | 14.4 | 10.6 | 13.8 | |
Table 6: Method efficiency of bosentan across all QC levels after extraction with different techniques and analyzed on different ion source in MS (n=6).
For the SPE process, Bond Elute Plexa cartridges were used, and various ratios of aqueous and organic and solvents were tried during the selection of washing and elution solvents. The detailed observation is given in Table 4. 100% water was selected as the washing solvent as the incorporation of the little amount of organic solvent (methanol) resulted in decreased ME. Pure methanol was used as the eluting solvent on the basis of highest recovery achieved. Washing and elution steps were repeated twice.
SPE technique was found to be the best technique in comparison to all other processes by ion suppression and ME. Our results are by earlier reported results that SPE is the technique of choice to increase ME and to eliminate matrix effects. The factor that could have led the higher ME in the SPE technique may be that we used water for washing step to get good recovery and sensitivity. Sample loss was avoided at this step as no organic solvent was included [4,26]. In LLE technique, the factors responsible for the slight decrease in ME could be attributed to the solubility of the internal matter of phospholipids and protein of the plasma in t-butyl methyl ether used in the extraction process.
The best results regarding matrix effect for all the three processes were observed on APCI ion source. A possible reason for observing the best results on APCI, as compared to ESI, could be the little lower sensitivity of APCI, and because of this, the internal components of the plasma and other external components responsible for matrix effect could not be ionized in the ion source. Other reason for differences in matrix effect could be likely due to the different ionization process, with APCI, as it is based on gas phase reactions and ESI mainly based on liquid-phase reactions. However, samples of PPT analyzed on APCI also showed the vast extent of matrix effect; that might be due to the use of a supernatant liquid that contained a higher concentration [4] of endogenous matrix components [12]. In PPT, the cleanup of biofluid is minimal due to the non-selective nature of the technique [3].
Inter and intra-day precision and accuracy of the analyte were performed after completion of the experiment to suitably use the developed method for the analysis purpose. The combination used was APCI-MS-MS with SPE extraction technique. The calibration curve in plasma was found to be linear from 10.04 ng /mL to 2002.42 ng/mL for BSN. Calibration curve was drawn using peak area ratio of analyte to the IS and by using linear, weighted least squares regression analysis with a weighting factor of 1/(x)2. The value of correlation coefficient (r) was found to be greater than 0.99 during analysis of precision and accuracy batches (Table 7). The Intra-day and inter-day accuracy ranged from 100.70-104.66%. The Intra-day and inter-day precision (% CV) ranged from 1.19-5.38.
| Solid Phase Extraction method | QC Sample Nominal Concentration (ng/mL) | ||||
|---|---|---|---|---|---|
| LOQQC (10.04) | LQC (25.37) | MQC (907.10) | HQC (1645.64) | ||
| % Accuracy | Intra day* | 102.39 | 103.88 | 101.46 | 101.34 |
| Inter day# | 104.66 | 104.40 | 100.70 | 100.92 | |
| % Precision | Intra day* | 4.61 | 3.64 | 1.34 | 1.94 |
| Inter day# | 5.38 | 3.74 | 1.19 | 1.62 | |
*n=12, Accuracy expressed as (mean calculated concentration/nominal concentration) × 100; #n=18, Precision or coefficient of variation (CV),expressed as the ratio of the standard deviation to the mean.
Table 7: Precision and Accuracy of QC Samples Analyzed on Ion Trap MS (APCI Source).
Our results demonstrated that matrix effect was dependent on ionization source type and sample preparation techniques. The matrix effect observed for both types of ionization sources, but the ESI was found to be more vulnerable in comparison to APCI. Sample preparation techniques were having a direct influence on matrix effect. Matrix suppression data indicated that SPE was the most efficient technique to avoid ion suppression followed by LLE and PPT respectively. LC-APCI-MS/MS was found to be the ionization of choice for quantitative analysis of BSN and other drugs with similar physicochemical properties. However, in order to develop an analytical method for a molecule with minimal matrix effect, selection of sample processing technique and selection of ionization source of particular sensitivity should be considered as key points.