Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2014) Volume 3, Issue 3
Though after a start of genome sequencing most of the protein sequences are deposited in databases, some
proteins remain to be annotated and functionally characterized. Analyzing and annotating the function of Hypothetical
proteins of Leptospira interrogans is very important which is a pathogenic spirochete causes various complications in
human and animals. Randomly we have selected 12 sequences of hypothetical proteins and were analyzed through
web tools as Pfam, CD Blast, Expasy's to determine physicochemical properties and protein family information based
on conserved domains. Proteins from families AdoMetDC, LRR, and PilZ are most conserved in many microorganisms
can be targeted to develop efficacious drug molecules. Ligand binding site and protein structure prediction was done by
using Qsite finder and PS2 server which will help to develops therapeutic molecules in docking studies. Present study
has shown intracellular interactions of proteins involved in drug resistance, transcription factors, and transport channels.
Keywords: Hypothetical protein; Leptospira interrogans; Bioinformatics tools; Functional analysis
Genome sequencing projects and genetic engineering has revealed many aspects of complex cellular environment containing large number of proteins. Despite sequences of most of organisms are available and proteins coded are studied experimentally, there are some proteins whose functions are unknown, need to be characterised [1]. Such proteins are known as Hypothetical Proteins (HP) sequences [2] of which are known but there is no evidence of experimental study [3]. There is extensive need to study and classify these hypothetical proteins which can open new way to design drug molecules against infectious organisms. Functional annotation of HPs involved in infection, drug resistance, and essential biosynthetic pathways is important for development of the potent antibacterial against infectious agents. Improved understanding of these proteins may make them potential targets of antimicrobial drugs [4]. Leptospira interrogans is gram negative spirochete, having an internal flagella is pathogenic which causes Leptospirosis [5-7], other serovars (strains) are distinguished on the basis of cell surface antigens. These are infectious to animals, but through animal urine can be spread to human [8]. Leptospira enters in body via broken skin, mucosa and spreads in body, if immune system fails to stop the growth of bacteria it cause severe hepatic and renal dysfunctions [9,10]. This present study highlights the in silico studies to characterize HPs from Leptospira interrogans.
Sequence retrieval
KEGG (Kyoto Encyclopedia of Gene and Genomes) is a large collection of databases having entries of genes, proteins, pathways in metabolism and diseases, drug and ligands of organism [11]. We have selected the Sequences of 12 hypothetical proteins of Leptospira interrogans randomly from KEGG database (www.genome.jp/kegg).Gene IDs for selected proteins are gi|24214908, gi|24215649, gi|24215664, gi|24215909, gi|24216444, gi|24217373, gi|24213620, gi|24214752, gi|24214753, gi|294827583, gi|294827687, gi|24213945.
Pfam
Pfam is curated Protein families database, it uses jackhmmer programme (HMMR3). To give profile HMM (Hiden Markov Model) with PSI-BLAST, which were searched against UniProt [12]. However, to include protein in a family its domain and sequence bit scores must be equal or above the Gathering Thresholds (GA). Pfam gives Pfam A families which are manually curated and Pfam B families generated automatically [13].
Batch CD search
Hypothetical Protein sequences were searched for conserved domains at batch CD search, which gives results by using MSA and 3D structures for homologous domains available on Pfam and SMART [12,14].
ExPASy-ProtParam tool
ProtParam tool (www.expasy.org/tools/protparam.html) was used to estimate physicochemical parameters of hypothetical proteins [15]. Query protein can be submitted in form of SWISS/TrEMBL ID or protein sequence. Server provides directly calculated values of pI/MW (Isoelectric point, Molecular Weight), Percentage of each amino acid, Extinction Coefficient (EC), Instability Index (II) [16], Aliphatic Index (AI) and GRAVY (Grand Average of Hydrophobicity).
SOSUI server
Amphiphilicity index and Hydropathy index of query protein sequences were calculated by SOSUI server which categorises protein into cytoplasmic or transmembrane nature [17].
Protein-Protein Interaction network
Protein in the cell environment interacts with other proteins, in silico these interactions were studied by STRING v9.1 (Search Tool for Retrieval of Interacting Genes). STRING is a large repository of protein-protein interactions involving functional interactions, stable complexes, and regulatory interactions among proteins [18,19]. Figure 1 Shows resulting protein-protein interaction network of selected hypothetical proteins, for better understanding interaction networks should be seen on server site.
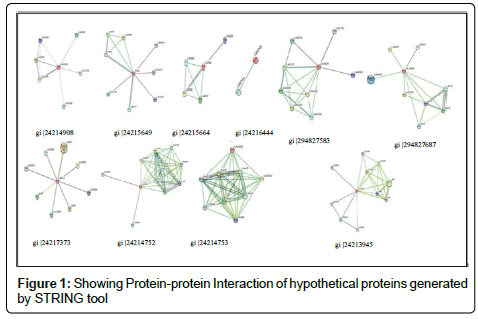
Figure 1: Showing Protein-protein Interaction of hypothetical proteins generated by STRING tool
Disulfide-Bonding in protein
Disulfide bonds among cysteine residues in protein plays an important role in folding it into functional and stable conformation. DISULFIND server utilizes SVM binary server to predict bonding state of cysteins, these cysteins are paired by Recursive Neural Network to show disulfide bridges [18]. Information about disulphide bonding helps in experimental structure determination and defining stability of the proteins.
Protein structure prediction
Protein structure prediction server (PS)2 [20] requires query sequence in fasta format to generate 3D structure by comparative modelling. Server utilizes consensus strategy to find template using PSI-BLAST and IMPALA. Query sequence and template aligned by T-coffee, PSI-BLAST, and IMPALA [17]. 3D structures are predicted from template using MODELLER and visualised by CHIME, Raster3D. Resulting 3D structural model of selected hypothetical proteins are shown in Figure 2.
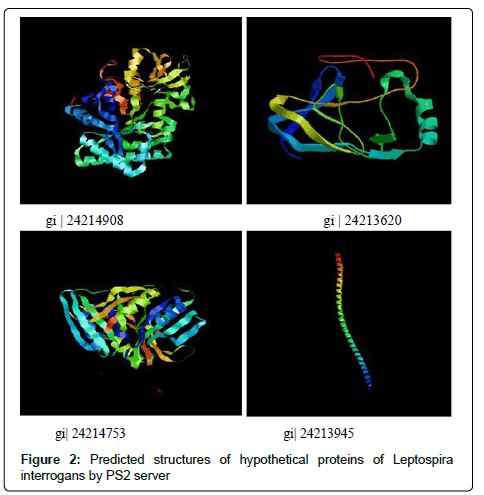
Figure 2: Predicted structures of hypothetical proteins of Leptospira interrogans by PS2 server
Ligand binding site prediction
Q-site finder server was used for binding site prediction in selected proteins [21]. Server uses energy based methods to find clefts on protein surface for ligands [22]. These hot spots for ligand binding have predicted after ranking their physicochemical properties as hydrophobicity, desolvation, electrostatic & van der waal potentials.
ProtParam tool computes different physicochemical parameters depending on the queries submitted to the databases. Isoelectric focusing separates proteins according to pI where pH gradients are developed [23]. Predicted pI via server may not be adequate because in case of high number of basic amino acids and lower buffer capacity. By using pH gradients and calculated pI, proteins can be separated experimentally. MW of proteins along with pI is used for the 2D gel electrophoresis. EC shows a light absorbed by a protein relative to their composition at a specific wavelength. EC given (Table 1) are calculated with reference to Tryptophan, Cysteine, Tyrosine [17]. Instability Index (II) refers to the stability of the protein in test tube [24]. Among studied proteins gi|24214908, gi|24215664, gi|24216444, gi|24213620, gi|24213945 were found to be unstable, and rest are stable (proteins with II above 40 are unstable). Aliphatic amino acid constitutes the aliphatic index (a relative volume of aliphatic side chains). Increased AI results into a hydrophobic interactions and thus gives thermostatic stability to protein, predicted AI and II shows inverse relation for stability except these two proteins gi|24215664 andgi|24215909. GRAVY [26] values are a ratio of all hydropathy values of amino acids to the number of residues in sequence. Smaller the GRAVY [25] more hydrophilic is protein, gi|24214908 and gi|24213945 proteins found the most hydrophilic. In case of 3D structure hydrophilic domains tends to be on exterior surface, while hydrophobic domains avoids external environment and forms internal core of the protein. Search of family for hypothetical proteins based on conserved domains having consensus sequence in their structure is given in Table 2. Hypothetical protein gi|24214908 found to be a member of GH18_CFLE_spore_hydrolase, Cortical fragment Lytic Enzyme bearing a catalytic domain from glycosylhydrolase, an enzyme used in breaking spore peptidoglycans so as to activate it for germination when favourable conditions are available. Hypothetical protein gi|24215649 from PDZ_serine_ protease involved in protein reassembly and work as a heat shock protein. Protein gi|24215664 belongs to Leucine-Rich Repeats (LRR), ribonuclease inhibitor like family. LRR are motifs having role in protein interactions in complex networks (Table 3). S-adenosylmethionine decarboxylase (AdoMetDC) enzyme for biosynthesis of spermine and spermidine by decarboxylation of SAM belongs to Ado_Met_dc family (gi|24217373). Pilz domain in gi|24213620 is found in bacterial cellulose synthase and other proteins that forms biofilm around a bacterium and involve in effluxing drug [26]. Hypothetical protein gi|294827583 (FecR superfamily) is involved in Iron transport system in bacterial membranes, Fe3+ (insoluble) loaded on citrate carrier is sensed by FecR protein found in periplasmic space in bacterial membrane [27]. Protein sites are predicted as cytoplasmic, host associated, extracellular, cytoplasmic membrane proteins (Table 4). SOSOI server predictions (Table 5) shows that positively charged amino acids are more at the end of transmembrane region. Analysis by DISULFIND server (Table 6) shows the Cysteine residues involved in disulphide bonding of hypothetical proteins. Protein-protein interaction study has shown some hypothetical proteins are involved in essential cellular process such as transport across membrane, biosynthesis of molecules, translational regulation. Hypothetical protein gi|24214908 (Figure 1) interacts with SUA5 protein which is known as one of translational regulator from YrdC/SUA5 family. Search for gi|24215909 shown to be involved in chloride transport with chloride channel protein (EriC gene). Protein gi|24217373 found to be interacted with S-layer like protein (slpM) which forms layer around bacteria to attach other surfaces and protect it from environment. Additionally it involves in cell dividing processes and transport across membrane. Protein gi|294827687 had shown interaction with proteins for bleomycin resistance, chorismatesynthase (Trp biosynthesis) and Mammalian Cell Entry (MCE) like proteins. Figure 2 shows 3D structures of proteins gi|24214908, gi|24213620, gi|24214753, gi|24213945 predicted from amino acid sequence on PS2 server by using templates 1vf8A, 3bo5A, 1f9zA, and c2efsA respectively.
| Sequence ID | No of AA | MW | pI | EC | II | AI | GRAVY |
|---|---|---|---|---|---|---|---|
| giǀ24214908 | 535 | 61891 | 8.27 | 100980 | 42.45 | 69.66 | -0.761 |
| giǀ24215649 | 445 | 50918 | 9.20 | 43445 | 39.23 | 79.26 | -0.396 |
| giǀ24215664 | 426 | 49102 | 8.7 | 13535 | 60.67 | 124.98 | -0.460 |
| giǀ24215909 | 251 | 26175.9 | 5.24 | 17795 | 28.55 | 65.58 | -0.161 |
| giǀ24216444 | 241 | 28062 | 8.74 | 11920 | 47.89 | 93.44 | -0.297 |
| giǀ24217373 | 288 | 32486.7 | 5.49 | 26275 | 36.72 | 83.30 | -0.267 |
| giǀ24213620 | 428 | 50351.8 | 5.13 | 31290 | 45.52 | 106.73 | -0.065 |
| giǀ24214752 | 399 | 46207.3 | 6.71 | 112550 | 34.23 | 121.93 | 0.631 |
| giǀ24214753 | 182 | 20886.2 | 8.6 | 19285 | 37.95 | 95.33 | -0.276 |
| giǀ294827583 | 309 | 33712.5 | 8.68 | 11710 | 36.34 | 88.61 | -0.358 |
| giǀ294827687 | 199 | 22228.3 | 9.62 | 13075 | 33.11 | 116.03 | 0.323 |
| gi|24213945 | 212 | 23671.6 | 3.90 | 1490 | 51.06 | 90.71 | -0.743 |
Table 1: Physicochemical properties of proteins using Protparam tool.
| Sequence ID | Family |
|---|---|
| giǀ24214908 | GH18_CFLE_spore_hydrolase,GH18_chitinase-like superfamily |
| giǀ24215649 | PDZ_serine_protease |
| giǀ24215664 | LRR_RI superfamily |
| giǀ24216444 | Macro superfamily |
| giǀ24217373 | AdoMet_dc superfamily, SET superfamily |
| giǀ24213620 | DUF1577 superfamily, PilZ superfamily |
| giǀ24214752 | DUF2157 superfamily |
| giǀ24214753 | GDYXXLXY superfamily |
| giǀ294827583 | FecR superfamily |
| giǀ294827687 | DUF2179 superfamily |
| gi|24213945 | DUF904 superfamily |
Table 2: Family distributions of HP’s by Batch CD-BLAST analysis.
| Protein | Site Volume(cubic A0) | Amino acid involved |
|---|---|---|
| giǀ24214908 | 548 | TYR 6, TRP 10, PHE 37, ALA 38, GLY 39,THR 48, GLY 76, GLY 77, TRP 78, LYS 79, PHE 80, ASP 117, GLN 119, TYR 120, THR 162, ALA 164, GLY 165, ILE 166, MET 189, TYR 191, ASP192, LEU 193. |
| giǀ24213620 | 465 | PRO 34, ALA 35, LYS 134, LYS 135, GLY 136, TRP 137, GLY 138, ASP 175, SER 176, ASN 177, TYR 178, ARG 206, PHE 207, LEU 208, ASN 209, HIS 210, SER 211. |
| giǀ24214753 | 201 | ARG 9, VAL 10, GLY 11, ARG 15, ASP 64, LYS 65, TYR 66, GLU 67, LEU 68, PRO 116, ASP 117, GLY 118, TYR 119. |
| gi|24213945 | 73 | ASN 42, ASP 43, GLN 44, LYS 46, LEU 47, GLU 50 |
Table 3: Prediction of Amino acids involved in Ligand binding (Q-site finder).
| Sequence ID | Protein Localization |
|---|---|
| giǀ24214908 | Unknown |
| giǀ24215649 | Extracellular |
| giǀ24215664 | Cytoplasmic |
| giǀ24216444 | Cytoplasmic |
| giǀ24217373 | Cytoplasmic |
| giǀ24213620 | Cytoplasmic Membrane |
| giǀ24214752 | Cytoplasmic |
| giǀ24214753 | Unknown |
| giǀ294827583 | Cytoplasmic Membrane |
| giǀ294827687 | Cytoplasmic |
| gi|24213945 | Unknown |
Table 4: Protein localisation by PSORTB.
| Gene ID | N- Terminal | Trans membrane region | C-Terminal | Type | Length |
|---|---|---|---|---|---|
| giǀ24215649 | 60 | FYFFKIFVLVLCSLTISVPLKAE | 82 | PRIMARY | 23 |
| 130 | AIVADQDFLLALLLPGEFPLFS | 109 | SECONDARY | 22 | |
| giǀ24215664 | 71 | LMILLILICFSCKLQAQ | 87 | PRIMARY | 17 |
| giǀ24214752 | 94 | YYSFLILGVVIIGIGVLAIIAAN | 116 | PRIMARY | 23 |
| 121 | DDLVKLGFSFIVLTFVAGLSFW | 142 | PRIMARY | 22 | |
| 149 | LFTVFIVLYSILILGMIGLISQV | 171 | PRIMARY | 23 | |
| 188 | LSCLFLITTDSKTFLHLWILGFQ | 210 | PRIMARY | 23 | |
| 303 | FSFPWVIMICRLLILTPIFYLLI | 325 | PRIMARY | 23 | |
| giǀ24214753 | 62 | KLILPIALVFPILFFVSEIITLE | 84 | PRIMARY | 23 |
| giǀ294827583 | 61 | YLSIVILCTFAMLLLVC | 77 | PRIMARY | 17 |
| giǀ294827687 | 73 | VLLPCFIFLSRVTDVSIGTIRVI | 95 | PRIMARY | 23 |
| giǀ294827687 | 103 | GIAASLGFLEVLLWVVVITQVIK | 125 | PRIMARY | 23 |
| giǀ294827687 | 138 | GGFATGTFIGMILEEKLAIGFSL | 160 | SECONDARY | 23 |
Table 5: SOSOI results for proteins.
| Sequence ID | Bonded Cysteines |
| giǀ24215909 | 55-210, 192-217, 200-236 |
| giǀ24214753 | 69-107, 87-118, 89-126 |
Table 6: Cysteine residues involved in disulphide bonding.
Development of potential bioinformatics tools and databases has opened new platform for in-silico study. Currently it is very needful to annotate and characterize hypothetical proteins in Leptospira interrogans serovar. These hypothetical proteins may have an imperative role in producing many virulence factors and cause serious infection or disease.We have analyzed 12 hypothetical proteins from KEGG database and categorized its physicochemical properties and recognized domains and families using various bioinformatics tools and databases. The structures were modeled and their ligand binding sites were identified.Physicochemical predictions made for hypothetical proteins, which can be used to find therapeutic agents against infections caused by Leptospira interrogans. Some of hypothetical proteins serves as channel proteins, ribosomal proteins or are involved in cell cycle process. Families which were identified for these hypothetical proteins are involved in normal cellular processes and the resistance against drugs. Ligand binding hotspots were found with Q-sitefinder which shown amino acids involved in interaction with ligands. It will help in study of molecular docking for development of potent and effective target against Leptospira infection.
This study was supported by NIPER Guwahati academic staff. We are very grateful for their excellent support in every manner.