Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 2
Functional analysis and interpretation of large-scale proteomics and gene expression data require effective use of bioinformatics tools and public knowledge resources coupled with expert-guided examination. An integrated bioinformatics approach was used to analyze cellular pathways in response to ionizing radiation. ATM, or mutated in ataxia-telangiectasia, a serine-threonine protein kinase, plays critical roles in radiation responses, including cell cycle arrest and DNA repair. We analyzed radiation responsive pathways based on 2D-gel/MS proteomics and microarray gene expression data from fibroblasts expressing wild type or mutant ATM gene. The analysis showed that metabolism was significantly affected by radiation in an ATM dependent manner. In particular, purine metabolic pathways were differentially changed in the two cell lines. The expression of ribonucleoside- diphosphate reductase subunit M2 (RRM2) was increased in ATM-wild type cells at both mRNA and protein levels, but no changes were detected in ATM-mutated cells. Increased expression of p53 was observed 30min after irradiation of the ATM-wild type cells. These results suggest that RRM2 is a downstream target of the ATM-p53 pathway that mediates radiation-induced DNA repair. We demonstrated that the integrated bioinformatics approach facilitated pathway analysis, hypothesis generation and target gene/protein identification.
Keywords: Bioinformatics, Proteomics, Radiation, Purine metabolism, DNA repair, Pathway and network
ATM: Ataxia Teleangiectasis Mutated; RRM2: Ribonucleotide Reductase subunit M2, or Ribonucleoside-diphosphate Reductase subunit M2; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; iProXpress: integrated Protein eXpression system; UniProt: Unified Protein Resource; 2D-gel/ MS: Two-dimensional gel/Mass Spectrometry; PIR: Protein Information Resource.
The last decade has seen a rapid expansion of genomics, transcriptomics, proteomics, and other omics studies applied to all areas of biomedical research. High-throughput technologies such as DNA microarray and mass spectrometry (MS)-based proteomics allow generation of large amounts of data from a single experiment. However, high-throughput data are generally of high variation, low reproducibility, noisy (von Mering and Bork, 2002), thus analysis and interpretation of the omics data remain challenging and require effective bioinformatics approaches. Biological interpretation of high-throughput data for forming hypotheses and for guiding experimental validation is typically a downstream process of the omics workflow after the high-throughput raw data are processed for functional analysis. At the core of functional interpretation of omics data is the knowledge (such as annotations and literature data) provided to the biological objects, being genes, mRNAs, or proteins from various molecular databases. Meanwhile bioinformatics tools have been developed for analyzing and interpreting the large lists of genes or proteins, such as DAVID (Huang et al., 2007), BABELOMICS (Al-Shahrour et al., 2005), Ingenuity (http://www.ingenuity.com/) and GeneGO (Ekins et al., 2007) for function and pathway analysis of large-scale data.
While bioinformatics tools have greatly assisted data analysis, a careful review of the major steps and flow of data in a typical high-throughput analysis reveals gaps that need to be addressed. One issue is the lack of standardization when dealing with a large list of proteins or genes annotated in different sources. For example, different protein IDs/names may be used for the same protein in different sources, even different versions of the same database may result in different IDs if the database identifier is not stable. The lack of standards presents a continuing challenge for integrating annotations from heterogeneous databases. Consequently, expression analysis is often carried out in an ad hoc manner, with a fragmented and inefficient use of rich annotations available in various resources. In addition, the effectiveness of the bioinformatics analysis system often relies on the amount and the type of knowledge available for genes and proteins annotated in the databases. To provide effective protein or gene ID mapping and comprehensive annotations for the largescale data analysis, we integrated two databases, UniProt (UniProt Consortium, 2008) and iProClass (Wu et al., 2004), into an integrated bioinformatics analysis system, iProXpress recently developed at the Protein Information Resource (PIR) (Huang et al., 2007). UniProt is a central international repository of protein sequences and functional information and provides the most comprehensive annotations for all proteins. iProClass database is a protein knowledge base providing value-added annotations integrated from over 90 molecular biology databases. iProClass coupled with UniProtKB became a data powerhouse of the iProXpress system, serving as a basic infrastructure for the omics data mapping and as a knowledge source for data analysis and interpretation.
In this paper, we describe an integrated bioinformatics approach for the gene expression and proteomics studies of human fibroblasts derived from patients with ataxia teleangiectasis (AT) who are sensitive to ionizing radiation-induced DNA damage. Radiation induces a myriad of cellular responses, including genotoxic stress signaling, cell cycle arrest, activation of a complex DNA repair machinery, and metabolic changes (Valerie, et al., 2007; Jeggo and Löbrich 2006; Spitz et al., 2004). ATM, or ataxia-telangiectasia mutated was first identified in AT patients in 1995 (Savitsky et al., 1995). ATM plays critical roles in radiation- induced responses (Kastan et al., 2001; Kurz and Lees- Miller, 2004), and has been identified as a potential target for novel radiosensitizers (Sarkaria and Eshleman, 2001; Ahmed and Li, 2007). For example, small molecule inhibitors of ATM or downstream signaling molecules (Kim et al., 1999; Jung and Dritschilo, 2001) may offer a strategy to sensitize tumors to the lethal effects of ionizing radiation while sparing normal tissues.
To identify ATM-mediated pathways underlying cellular responses to ionizing radiation that lead to radiation resistance or sensitivity in cells, the AT-patient derived fibroblasts expressing mutated ATM genes or wild-type ATM were used as models. The two cell lines were subjected to proteomics and microarray experiments, analyzed by global expression profiling and pathway/ network analysis. We showed radiation-induced and ATMmediated major biological pathways and proposed proteins for further validation.
Experimental Data Source
The proteomics and gene expression data were obtained from radiation-treated AT5BIVA and ATCL8 cell lines. AT5BIVA was derived from human fibroblasts of ataxia teleangiectasis (AT) patient with mutated ATM (AT mutated) gene (Jung et al., 1995), while ATCL8 was derived by reintroducing the wild-type ATM gene into the AT5BIVA cells. The two cell lines were exposed to 10 Gy of ionizing radiation and analyzed at time intervals from 30 minutes to 24 hours. The proteomics data were obtained from two-dimensional gel electrophoresis (2D-gel) followed by MALDI-MS of the excised gel spots. The gene expression data were obtained using Affymetrix DNA microarray (U133A probe set of 14500 human genes) chip assays. The experimental procedures for cell culture and radiation treatment, 2D-gel, MALDIMS proteomics and microarray have been described elsewhere (Lee et al., 2001; Mewani et al., 2006). Protein identification from MALDI-MS was based on MASCOT search engine using UniProtKB/Swiss-Prot database. Lists of proteins were identified (with UniProtKB accession #) from differentially changed 2D-gel spots based on >=2-fold changes (p-value <=0.05), increased (including newly appeared spots after irradiation) or decreased (including spots only in control but disappeared after irradiation) for each time point and cell type. Lists of genes were identified (with Entrez Gene #) from differentially expressed mRNAs (increased or decreased) in microarray based on >= 1.5- fold changes (p-value <= 0.05).
Data Integration and Bioinformatics Analysis
We applied an integrated bioinformatics approach for the proteomics and gene expression data analysis. The iProXpress integrated protein expression analysis system (http:// pir.georgetown.edu/iproxpress/) was primarily used as a platform for the functional data analysis, coupled with the Ingenuity Pathway Analysis (IPA) tool for pathway and network analysis. A prototype of the iProXpress system has been applied to several previous high-throughput studies (Li et al., 2004; Chi et al., 2006; Hu et al., 2007). Below we briefly describe the bioinformatics analysis procedures.
Protein mapping: Gene or protein lists were mapped to UniProtKB protein entries primarily based on gene/protein identifiers. Genes with common identifiers such as GenBank, UniGene or Entrez Gene are mapped based on the PIR ID mapping service (http://pir.georgetown.edu/pirwww/search/ idmapping.shtml). For genes with no ID match, the mapping is based on sequence comparison, or name mapping if the sequence is not available. The protein and gene lists from AT5BIVA and ATCL8 cells were integrated into the iProXpress system after protein mapping.
Protein annotation: After protein mapping, rich annotations are described in a protein information matrix that captures salient features of proteins, such as functions and pathways, for given experimental data sets. These rich annotations are derived from comprehensive protein information that have been integrated into the UniProt and iProClass databases and from sequence analysis for homology-based inference.
Functional profiling: The gene and protein lists were divided into experimental groups based on cell types and time course for functional profiling using various functional attributes (i.e. annotation fields of the protein information matrix). Primarily used for functional profiling were GO slims (a subset of GO with high level terms at GO hierarchy) (http://www.geneontology.org/ GO.slims) and pathway information (e.g. from KEGG database).
Pathway and network analysis: Pathway visualization was based on pathway diagrams provided in source pathway databases such as KEGG and the IPA tool. An ATM protein interaction pathway map was also used, which was curated by scientists who initially discovered the ATM gene (Savitsky et al., 1995) and reflects the current state of knowledge for ATM-mediated pathways (available at http://www.cs.tau.ac.il/~spike/images/ 1.png). Network analysis was done using the IPA tool, which dynamically generates functional association networks based on curated literature information of protein-protein interaction, coexpression, and genetic regulation.
Figure 1 depicts genotypes the overview of an integrated bioinformatics approach to analyze and interpret the proteomics and gene expression data from irradiated cells with mutant or wild type ATM genotypes.

Figure 1: Integrated bioinformatics approach for radiation-induced function and pathway analysis from proteomics and gene expression data.
Figure 2 shows the iProXpress web interface for searching, browsing, and profiling the experimental groups of different cell types, time courses, and protein or mRNA level changes. The interactive graphical user interface provided several functionalities for data analysis, such as selecting data groups, browsing the proteins and associated annotations, and expression profiling using GO slims and pathways.

Figure 2: iProXpress web interface for browsing and profiling proteomics and microarray data.
1) Selected experimental groups can be chosen from the pull down menu. 2) Boolean operations can be used to query the data, such as “get all proteins identified from proteomics (A_8_30m*) OR microarray (B_8_30m*) in ATCL8 cell at 30 minutes.” 3) The “protein information matrix” displays protein list from the selected groups, and provides annotations integrated from over 90 sources. 4) The “Display Option” allows selection of desired fields for display. 5) For the given list of proteins, the interface provides functional profiling “buttons” to show profiles in GO slim (molecular function, cellular component, and biological process) or KEGG pathways. 6) The interface also provides protein sequence analysis tools listed such as BLAST, FASTA and sequence alignment. 7) An example of GO biological process profile. 8) Comparative profiling across selected data groups based on given GO categories.
Radiation-Induced Changes in Expression Profiles
The 2D-gel/MS proteomics and DNA microarray data generated from radiation-treated AT5BIVA and ATCL8 cells are summarized in Table 1, which shows total numbers of UniProt protein entries mapped from proteomics and gene expression data. Most up-regulated proteins were observed at 3hr post-irradiation in both AT5BIVA and ATCL8 cells, and with many more up-regulated in ATCL8 than in AT5BIVA cells. In contrast, most downregulated proteins were seen at 30min in ATCL8 and at 24hr in AT5BIVA cells. At gene expression level, prominent responses to radiation at early time in ATCL8 cells were observed, for example, three times as many mRNAs were up-regulated at 30min in ATCL8 (33) as in AT5BIVA (11) cells, while up-regulation of most genes was only seen 1hr after radiation in AT5BIVA cells. These differences showed that ATCL8 was more radiation-responsive at both protein and mRNA levels at earlier time than the ATM-mutated AT5BIVA cells. Compared to AT5BIVA, ATCL8 cells were shown to quickly respond to irradiation at 30min by increasing more gene expressions and by decreasing the amounts and/or activities (presumably modification states) of more proteins, followed by increasing more at 3hr.
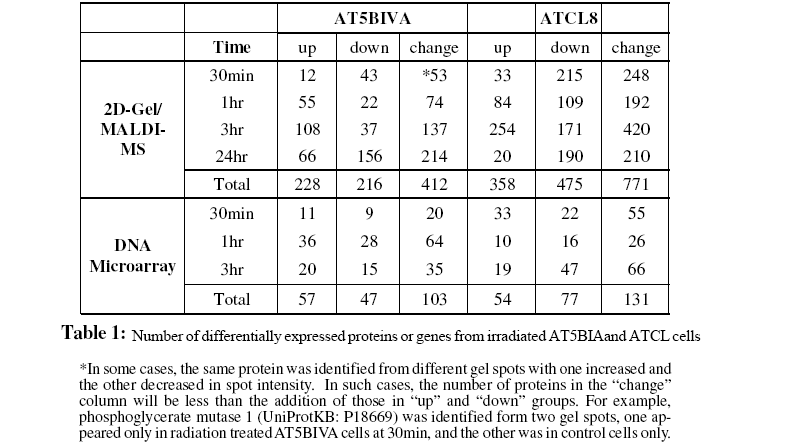
The profiling of the differentially changed proteins or genes from irradiated cells based on GO slims and the KEGG pathways provided global views of functional changes in these cells. Table 2 shows the major GO biological process categories of radiation induced protein changes. The total changed proteins (combined up- and down-) in the two cell lines generally showed similar profiles among top categories of GO biological processes. However, profiles based on up- or down-regulated proteins showed clear differences between the two cell lines. For example, in AT5BIVA cells, a higher percentage of proteins involved in cell cycle was down-regulated (8.3%) than up-regulated (4.8%), and more were up-regulated than down-regulated in RNA metabolism, transcription, and protein biosynthesis. In ATCL8 cells, a higher percentage of proteins were up-regulated in signal transduction and protein modification, while more were down-regulated in protein biosynthesis.
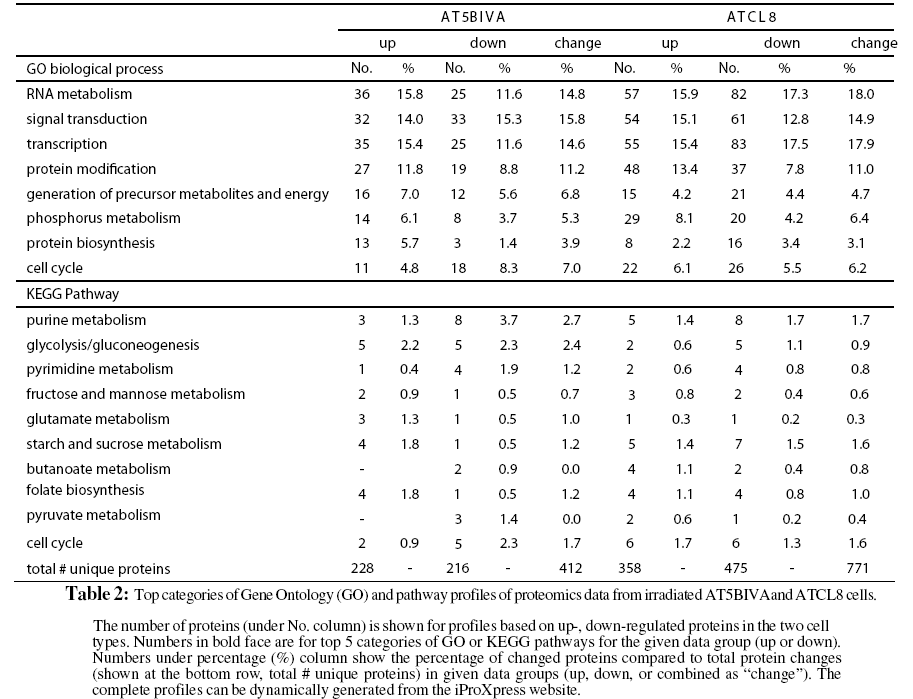
When profiling is performed using KEGG pathways for the total changed proteins, differences were observed in the percentages of proteins involved in purine metabolism, glycolysis/gluconeo-genesis, pyrimidine metabolism, and glutamate metabolism in the two cell lines. Pathway profiling based on the up- or downregulated proteins resulted in more differences between the AT5BIVA and ATCL8 cells. For example, higher percentages of down-regulated proteins in purine metabolism and of up-regulated proteins in starch and sucrose metabolism and folate biosynthesis were observed in AT5BIVA cells. Also consistent with GO process profiles, more cell cycle proteins were seen downregulated in AT5BIVA while more were up-regulated in ATCL8 cells. Overall, metabolic pathways were clearly affected, and purine metabolism was the most affected pathway in irradiated AT5BIVA and ATCL8 cells based on the expression profiling using iProXpress as well as from the Ingenuity pathway profiles (not shown).
Biological Pathways and Signaling Proteins in Response to Radiation
Although the general profiles in Table 2 provided global views of major functional changes in the two cell lines without regard to specific time points, profiles based on more specific or focused data groups, such as at certain time points, offered more biological insights. We selected a proteomics data set at 3hr from both AT5BIVA and ATCL8 cells and a microarray data set at 30min from ATCL8 only for further analysis, when most differentially changed protein or gene expressions were observed or most upregulation of proteins or genes occurred (Table 1). The comparative pathways profiling of four data groups representing the upand down-regulated proteins from AT5BIVA and ATCL8 cells at 3hr post-irradiation showed that purine metabolism is the most predominant pathway with 10 differentially expressed proteins, and major differences exist between the four data groups (Figure 3).

Figure 3: Comparative pathway profiling of proteomics data from AT5BIVA and A TCL8 cells at 3hr post-irradiation.
Table 3 lists proteins of purine metabolism from all time points (30min to 24hr) in AT5BIVA and ATCL8 cells. Most enzyme changes in this pathway occurred at 3hr in both cell lines, and those changed at other time points were mostly down-regulated in both cells. Strikingly, while most changed enzymes were downregulated at 3hr in AT5BIVA cells, all changed enzymes were up-regulated at 3hr in ATCL8 cells. Two enzymes with opposite changes were identifiet from the two cell lines, adenylate kinase 2 (up in ATCl8 and down in AT5Cl8 at 30min), and IMP dehdrogenase 2 (up in ATCl8 at 3hr and down in AT5BIVA at 24hr).
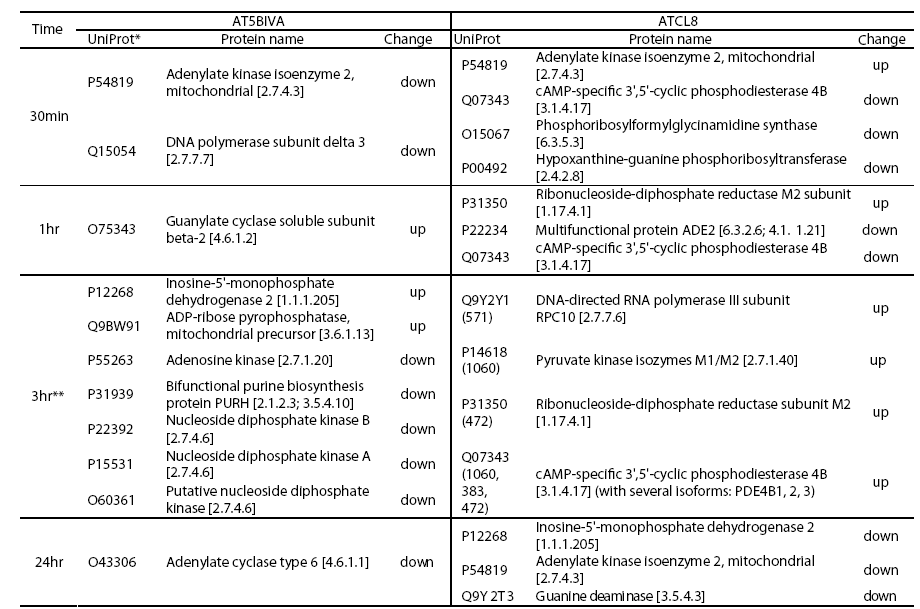
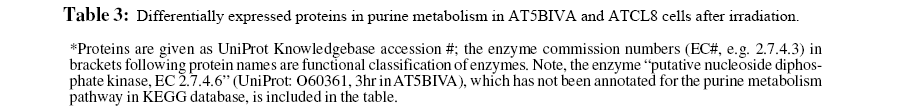

Figure 4: Differentially expressed enzymes in purine metabolism identified from irradiated AT5BIVA and ATCL8 cells. Enzyme Commission numbers (EC#, e.g. 1.17.4.1) are used to represent enzymes in metabolism. Highlighted in green background are known human enzymes annotated in the KEGG database. Differentially expressed enzymes in purine metabolism (Table 3) are superimposed onto this pathway diagram: blue-boxed are enzymes changed in AT5BIVA cells, red-boxed those in ATCL8 cells, and pinkboxed those from both cells. Areas circled with broken lines highlight closely related biochemical steps surrounding ADP/ATP (left) or GDP/GTP (right) metabolisms, which include most of these differentially expressed enzymes from either cell type.
Because of the relatively low numbers of differentially expressed genes from the microarray experiment, expressing profiling using GO or KEGG pathways was usually not revealing for most of the experimental groups (Table 1). Instead we focused on the differentially expressed genes from ATCL8 cells at 30min postirradiation, when more genes were differentially expressed in ATCL8 than in AT5BIVA cells, and most up-regulated genes in ATCL8 occurred. Table 4 lists gene products from the top 3 GO biological process categories, signal transduction, protein modification, and transcription, from the microarray experiment. Among them, p53, BRCA1 and HDAC1 were all up-regulated at 30min in ATCL8 cells and are also well-known to be involved in DNA repair and cell cycle control.
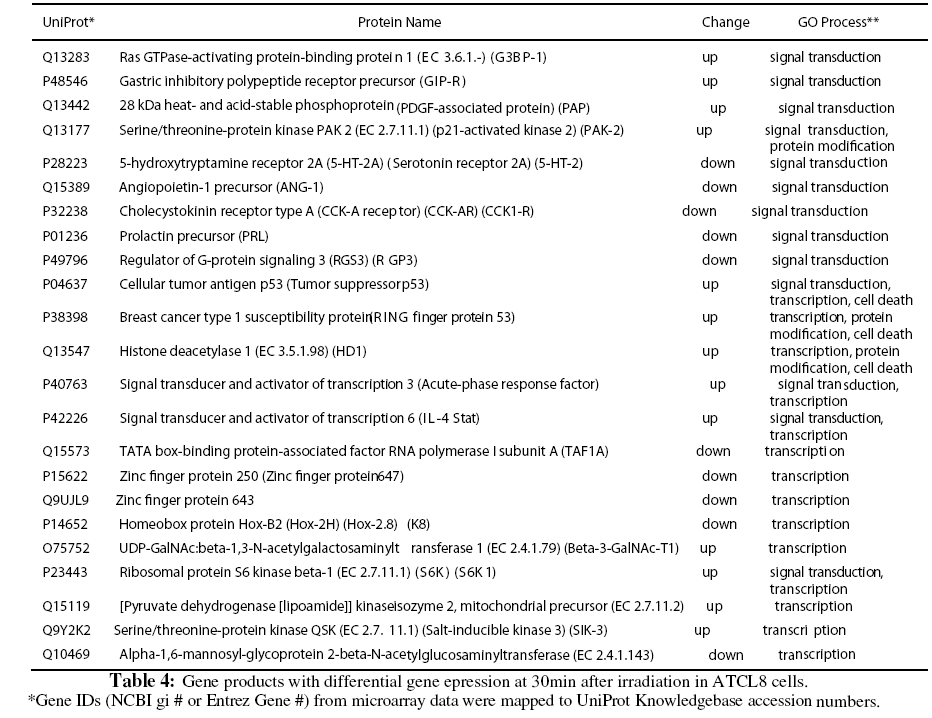
Furthermore, despite the low numbers of differentially expressed genes from microarray experiment, it was interesting to correlate these genes with differentially changed proteins from proteomics data. A total of 103 proteins (UniProt entries) from AT5BIVA and 131 from ATCL8 cells were mapped from from the microarray data of both AT5BIVA and ATCL8 cells (Table 1). Table 5 shows the common protein set of 13 proteins, namely the overlapping genes/proteins between the proteomics and microarray data, 10 were from ATCL8 and 3 from AT5BIVA cells.
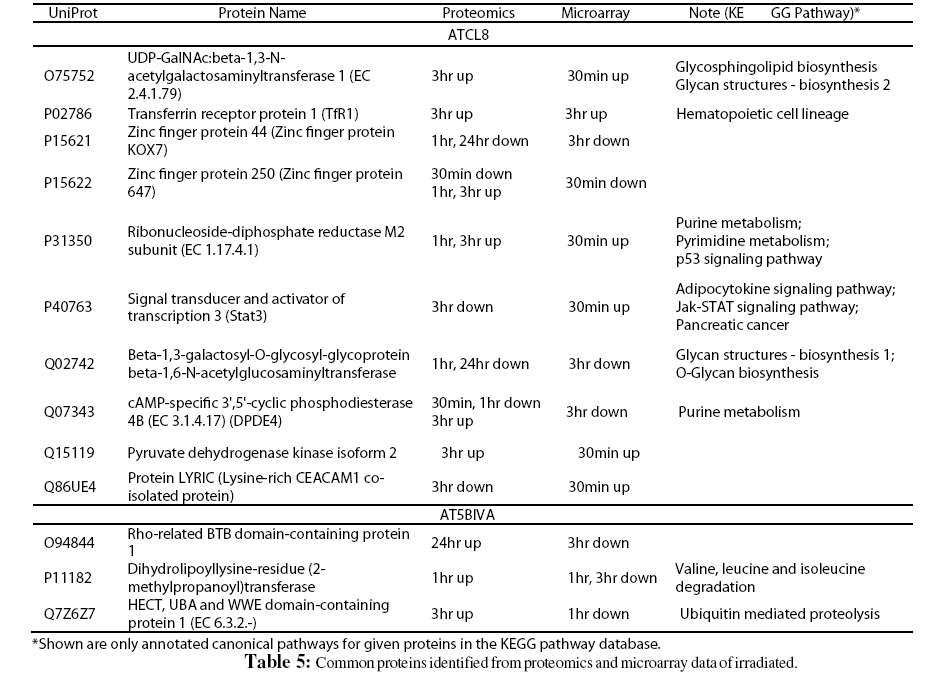
Interestingly, from above proteomics and microarray data RRM2 was shown to be increased at both mRNA and protein levels in ATCL8 cells, with mRNA level increased at 30min, and protein level increased at 1hr and 3hr (Tables 3 and 5, and Figure 4).
RRM2-Associated Functional Networks and Pathways
A critical rate-limiting enzyme in DNA synthesis, RRM2 expression increased in ATCL8 cells at 1hr and 3hr after irradiation, while no changes were detected in AT5BIVA cells. Since RRM2 is involved in DNA repair, we wanted to examine the functional association networks involving RRM2 in the context of current proteomic and gene expression data.
Figure 5 shows the network in which RRM2 is connected with several major DNA repair and cell cycle proteins, including HDAC1, p53, BRCA1, and CDKN2A, directly or indirectly. Except for CDKN2A, a negative regulator of cell cycle progression, the other three proteins were all differentially regulated in ATCL8 cells, suggesting that RRM2 play an important role in radiation-induced and ATM-mediated DNA repair processes and cell cycle control.

Figure 5: Functional networks showing RRM2 connected to other major DNA repair and cell cycle proteins, such as p53, BRCA1, HDAC1.
Since AT5BIVA and ATCL8 cell lines were specifically designed as models for examining ATM-mediated pathways, we used an ATM protein interaction pathway map to examine changed proteins or genes from proteomics and gene expression data. This pathway map (Figure 6, left) shows that two proteins directly interacted with and activated by ATM are p53 and BRCA1, which were up-regulated in ATCL8 cells. Based on the expression data and the network analysis, we hypothesize that RRM2 is involved in radiation induced ATM-p53-mediated DNA repair pathway in the ATCL8 cells (Figure 6, right). RRM2 directly binds p53 and upon irradiation dimerizes with RRM2 to form the ribonucleotide reductase (RR) holoenzyme complex. Increased RR activity will result in an increase in the pool of deoxyribonucleotide precursors for DNA synthesis which is required for DNA repair in response to radiation damage.

Figure 6: RRM2 is involved in radiation-induced ATM-p53-mediated DNA repair pathway.
In this study we used an integrated bioinformatics approach (Figure 1) to analyze and interpret the proteomics and gene expression data from radiation treated cells with mutant or wild type ATM genotypes. The iProXpress system provides a protein-centric data integration for functional analysis and allows direct comparison of different molecules (mRNA vs. protein) from same samples under study. As functional understanding of the omics data is underpinned by the current knowledge annotated in databases for given lists of genes or proteins from high throughput experiments, it is crucial to maximize the use of known knowledge from heterogeneous databases and resources. The iProXpress system uses both iProClass and UniProtKB databases for data mapping, data analysis and interpretation, and also takes advantage of the extensive informatics infrastructure at PIR, e.g. the Text Search engine for data browsing and searching. One of the most useful features of the iProXpress system is to allow comparison of functional profiles across multiple data sets or groups obtained from different issue/cell types and time points, or from different omics experiments. In particular, while differential profiles with GO slim or pathway terms may not be evident when generated from combined data groups, profiles from more specific groups may reveal clearer differences. For example, purine metabolism became evident when examining individual time points from both AT5BIVA and ATCL8 cells.
While existing annotations from databases are critical for the omics analysis, the knowledge base is still limited. GO has become a common standard for annotation and functional analysis, but currently only about half of all human genes/proteins are annotated with GO terms, and even less with experimentally validated and manually annotated GO functions. Compared to GO profiling, pathway and network mapping provide more biological insight, however, an estimated <10% of human genes/ proteins have been annotated with biological pathways in databases. Therefore, as part of the integrated bioinformatics approach, expert-guided analysis should be coupled with review of scientific literature for functional interpretation of the large scale omics data and for formulation of scientific hypothesis.
The expression profiling and pathway/network analyses have shown that enzymes of purine metabolisms, especially surrounding steps of the ADP/ATP and GDP/GTP synthesis, were differentially affected in irradiated AT5BIVA and ATCL8 cells. RRM2 is a small subunit of the RR complex that is well known for its role in DNA synthesis. RR is the only enzyme responsible for the reduction of ribonucleotides to their corresponding deoxyribonucleotides, providing a balanced supply of precursors for DNA synthesis and repair. It has been shown that an increase in RRM2 protein levels and RR activity in human nasopharyngeal cancer cells results in ionizing radiation resistance, which appears mediated by enhanced ionizing radiation damage repair during G2 phase of the cell cycle. However, overexpression of the large subunit, RRM1, of RR in these cells did not affect RR activity or ionizing radiation response (Kuo et al., 2003). RRM2 over expression is also associated with gemcitabine chemoresistance in pancreatic adenocarcinoma cells, and that suppression of RRM2 expression using RNA interference enhances gemcitabine-induced cytotoxicity in vitro (Duxbury et al., 2004). Human RRM2 has been shown to be a target of p53 through direct protein-protein interaction that leads to the nuclear accumulation of RR subunits after UV exposure (Xue et al., 2003), and inhibition of RRM2 by hydroxyurea results in increased sensitivity to UV irradiation in prostate cancer (PC3) cells (Zhou et al., 2003). Our results suggest that RRM2 is involved in the ATM- and p53-mediated signaling pathway leading to DNA repair in response to radiation in ATCL8 cells, while the ATM-mutated AT5BIVA cells became more sensitive to radiation possibly due to the impaired activation of RRM2 expression.
Most of proteins in this study were derived from the 2D-gel/MS experiment, and not all identified proteins from given 2D-gel spots were responsible for the observed changes. We used this integrated bioinformatics to help rational selection of candidate proteins for validation. Based on common pathways (e.g. purine metabolism) and their differential expression patterns, we can preferentially select those proteins commonly associated with a pathway over those not associated with the pathway for validation. Indeed, the enzyme RRM2, identified from a spot with 40 identified proteins at 3hr and a spot with 12 identifications at 1hr in ATCl8 cells (not shown), was actually one that was most likely to have changed, also consistent with the finding that RRM2 mRNA was up-regulated at 1hr in the same cells.
It was noted that the intersection between changed proteins and genes from the proteomics and gene expression data in this study was small. The lack of direct correlation between changes in proteins and genes from gene expression and proteomics experiments has been previously observed (Jansen et al., 2002; Hewick et al., 2003). This is due in part to the experimental artifacts and in part to differential post-transcriptional or post-translational regulation of genes or proteins. For example, an increased or new 2D-gel spot may result from increased protein phosphorylation without corresponding mRNA changes.Constructing gene regulatory networks may potentially help identify correlations between proteomics and gene expression data when direct correlation between the two is not apparent (Perco et al., 2005).
Besides identifying RRM2 as a potential downstream target of the ATM-p53-mediated pathway for DNA repair in response to radiation, other enzymes in purine metabolism and several other metabolic pathways, such as AK2, IMPDH, and NDK, were differentially expressed as well in the two cell lines. Interestingly, three forms of NDKs (nucleoside diphosphate kinase) were observed to be down-regulated in AT5BIVA cells. NDKs have recently been found to have DNA binding and exonuclease activities (Yoon et al., 2005). It is not clear however whether this is related to the reduced DNA damage repair in ATM mutated cells. Their roles and significance of these metabolic enzymes in the ATM-mediated pathways and in radiation responses remain to be further examined. Currently we are extending this study by applying metabolomics measurement to the two cell lines after irradiation, aiming to identify changes in metabolites in response to irradiation and the anticipated differential patterns in wildtype ATM vs. mutant ATM-expressing cells. Our current proteomics and gene expression data will provide a valuable reference for future analysis and interpretation of radiation damage- induced metabolites. We envision that integration and correlation of proteomics, functional genomics and metabolomics data generated from the same experimental system will provide new biological insight.
In conclusion, we have demonstrated an integrated bioinformatics approach that includes expert-guided examination of data to define radiation-induced and ATM-mediated pathways in cell models with wild-type or mutant ATM genotype. We have shown that purine metabolic pathways were differentially affected in response to radiation, and that RRM 2 was up-regulated only in ATM-wild type but not in ATM-mutated cells. We hypothesize that in this cell model, ionizing radiation activates ATM-p53- mediated pathway that directly targets RRM2 and leads to DNA damage repair, thus increasing radiation resistance in the ATCL8 cells
This work is supported in part by NIH/NCI grant (P01CA074175). The bioinformatics infrastructure for this study was supported in part by NIH grant U01-HG02712.