Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2021)Volume 10, Issue 1
Density functional theory; Molecular docking; Molecular dynamics simulations and Natural bond orbital; Drug action
DNA minor groove is the target for many anticancer and antitumor drugs. The forces that are involved in DNA-minor groove binding are electrostatic, van der Waal’s, hydrophobic, and hydrogen bonding [1]. Sequence specificity is important key tool for the drug-DNA interaction. Another important factor for the interaction is that the small molecule has a crescent shape which is complementary to the minor groove of DNA and thus minor groove binding is favored [2]. Frequently, minor groove shows selectivity towards AT region, several factors are responsible for this, first one is the electrostatic potential of AT-rich region is higher than that of GC-filled ones and another one is that the AT-rich grooves are narrower and deeper than GC ones [3]. DNA minor groove with alternating A and T, allows favorable van der Waal’s contacts between the drug and DNA instead of GC-rich region where the geometry of groove is altered by bulky amino groups of guanine bases [4,5].
In the current study, some different class of minor groove binder's (2,4-bis(4-amidinophenyl) furans and reversed diamidino 2,5-diarylfurans), labeled as mol-1, mol-2 mol-3 and mol-4 are taken, followed by a DNA sequence (PDB Id: 4AH0). Molecular modeling techniques such as geometry optimization, molecular docking and molecular dynamics simulation were used for the current research.
The ligands were first geometrically optimised in order for them to attain a minimum potential, as this would lead them to have electrostatically optimized chemical structure. Molecular Docking is simplest computational representation of DNA- ligand interactions [6]. After geometry optimization, ligands were docked to the selected DNA sequence for the identification of possible binding site followed by corresponding site binding energy was also obtained.
Various literatures have reported that, the minimum the site binding energy, the better the corresponding docked pose [7]. After geometry optimization and docking, the drug-DNA complexes were put to molecular dynamics simulation for 5ns and was observed that convergence is achieved within 5ns simulation. Obtained results not only predicted the conformational stability of the DNA with the ligands having had inserted between its base pairs but also gave a firm affirmation regarding the time dependence of the interaction and stability of drug-DNA complexes [8].
This article ends on a note that the three out of the four selected ligands had formed stable complexes with DNA; none of them had ruptured the DNA double helix over 5ns time scale simulation; interactions between drug and DNA was strong and significant; and helps in enriching the computational studies of drug-DNA interactions database and also paves ways for further sophisticated research viz, QM and QM/MM calculations [9,10]. The extension of such studies involving computational techniques is not only limited to drug design and molecular modelling but also to drug metabolism involving enzymes and their involved catalytic mechanisms [11].
System selection and preparation
The lead molecules (mol-1, mol-2, mol-3 and mol-4) were selected from literature [12]. Their chemical structures are shown in Figure 1. The DNA sequence; 4AH0 [13] was obtained from Protein Data Bank [14]. Its structural data including its specific nucleic acid bases sequences and PDB Id. is mentioned in Table 1 as obtained from Discovery Studio software package [15]. Water molecules and other residues, from selected DNA sequence were removed using UCSF Chimera before the start of docking calculations [16].

Figure 1: Chemical structures of the selected ligands.
| S. No. | PDB Id. | Nucleic Acid Sequence from Available Structure | Basic Information for Molecule |
|---|---|---|---|
| 1 | 4AH0 | B-DNA DODECAMER (5'-D(CGCAAATTTGCG)-3') | Cell Space Group: 19 (P212121 Origin-1 Choice: 1) |
| Chain A: CGCAAATTTGCG | Crystallographic Resolution: 1.20Å | ||
| Molecular Weight of Nucleic Acid Chains: 7268.84 | |||
| Chain B: CGCAAATTTGCG | Number of Nucleic Acids: 24 | ||
| Experimental pH: 6.5 |
Table 1: Nucleic Acid Report for 4AH0 as obtained from Discovery Studio Software.
Molecular geometry optimization
Using optimized ligand geometry before docking and other computational calculations is of utmost importance. The reason behind this is that geometry optimization would put the entire system in the lowest possible energy state; having least steric hindrances and charge. Further, the electrostatic charges also tend to get well distributed throughout the surface of the system leading to the electron transfer between definite pair of atoms; and eventually no distortions in the chemical structures are offered [17]. Here, the geometry optimization of selected ligands was carried out using Gaussian 09 software [18] using B3LYP hybrid functional at 6-31G** level of theory for their potentials to attain a local minima. These optimized ligands were then docked to the selected DNA sequence. Frequency calculation at the above level characterize the obtained point as minima on the potential energy surface in the optimize geometries.
Molecular docking studies
Molecular Docking is a widely used computational technique to predict the binding pocket of potent inhibiters in the vicinity of target molecules. The ability to determine the binding affinity of protein-ligand complexes is the key objective of computational docking [19]. Here, molecular docking was carried out using Autodock 4 software [20]. Docking calculations were set up using Lamarckian Genetic Algorithm (LGA). Gasteiger charges were added to each drug-DNA complex using Autodock Tools (ADT) before starting the docking simulations. A grid box, having various dimensions along the three coordinate axes, was prepared for each drug-DNA complex that enclosed the macromolecule. A 20 LGA run with a maximum cycle of 2500000 energy evaluations was performed for each of the drug-DNA complex. The docked pose with the least binding affinity was extracted and aligned with the receptor (DNA) for further analysis.
Molecular dynamics simulation
Molecular Dynamics is computational technique that is to study the dynamic characteristics, viz, protein folding, unwinding and other conformational changes including complex stability, over a specified period of time. Its applications have gained significant importance due to lack of experimental resources [21]. In the present work, GROMACS 5.0.4 software package [22] was used to carry out the molecular dynamics simulations. A total of four drug-DNA complexes were generated, after docking for molecular dynamics simulations viz, (4AH0+mol-1, 4AH0+mol-2, 4AH0+mol-3 and 4AH0+mol-4). These drug-DNA complexes were put to for 5000ps time scale simulation. There are many studies for the comparison of force fields for the nucleic acids but AMBER force fields [23] seems to be good for nucleic acid simulation due to the presence of specific topologies for the terminal nucleotides. Amber 03 force field already embedded in GROMACS software suite was used to generate the topology for the selected DNA sequence and antechamber module of AMBER program was used to generate topology of selected ligands through a python script: ‘acpype.py’ [24].The Ligand-DNA complex was solvated in a box of varying dimensions using TIP3P water model at 298K [25]. Sodium ions were then added to the solvated box containing the DNA-ligand complex by randomly replacing the water molecules in order to neutralize the system. Particle Mesh Ewald (PME) was used to handle long-range electrostatic interactions in periodic boundary conditions [26]. Energy minimization of the whole system was carried out in 25000 steps using Steepest Descent leap- Frog Integration Method followed by NVT ensemble equilibration at a constant temperature of 300K for 50s using Berendsen thermostat [27]. The system was then equilibrated with NPT ensemble at a constant pressure of 1atm in 25000 steps using steepest descent leap-frog integrator [27]. Particle Mesh Ewald (PME) was used to handle long-range electrostatic interactions in periodic boundary conditions and all the bonds involving hydrogen atoms were constrained using the LINCS algorithm [28]. Graphs were plotted using XMgrace software [29].
Natural bond orbital analysis
NBO analysis played the role of intermolecular orbital interaction in the complex, particularly charge transfer in the given wave function into localized form, corresponding to the one-center (lone pairs) and two-center (bonds) elements of the chemist's Lewis structure picture. In NBO analysis, the input atomic orbital basis set is transformed via natural atomic orbitals (NAOs) and natural hybrid orbitals (NHOs) into natural bond orbitals (NBOs). The NBOs obtained in this fashion correspond to the widely used Lewis picture, in which two-center bonds and lone pairs are localized. This is carried out by considering all possible interactions between filled donor NBOs and empty acceptor NBOs and estimating their energetic importance by second-order perturbation theory. For each donor NBO (i) and acceptor NBO (j), the hyper energy conjugated interaction (stabilization gained energy E(2) in kcal/mol) associated with electron delocalization between donor NBOs and acceptor NBOs is estimated as
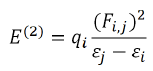
Where qi is the orbital occupancy, εi, εj are diagonal elements and Fi,j is the off-diagonal NBO Fock matrix element [30].
Present study was aimed to identify new leads, targeting binding affinity, structural stability and applicability for antimicrobial ligands with DNA. The results obtained through various computational calculations are discussed and summarized as follows:
Molecular geometry optimization
The geometry of the lead molecules needs to be optimized before docking so that the repulsions between the nearest bonded atoms, between their angles and between their dihedrals could get minimized and the lead molecules would attain a state having least repulsion and hence owing to maximum stability and correspond to minimum energy [30]. The optimized geometries of the selected ligands are shown below in Figure 2.

Figure 2: Optimized structures of the selected ligands.
Molecular docking
The selected possess of ligands (mol-1, mol-2, mol-3 & mol-4) for antimicrobial tendencies were docked to 4AH0 in search of best docked posed complex leading to stability. The docking results, corresponding to the selected DNA sequences are summarized below in Table 2. Docking calculations revealed that 4AH0 with mol-4 had least binding energy of -12.39 kcal/mol. This indicates that since mol-4 had lesser binding energy, it owes to maximum stability of the formed complex. The docked pose corresponding to each drug-DNA complex is shown in Figure 3. These figures reveal that all the four ligands bonded themselves to the minor groove of each DNA sequence. Docking calculations also revealed that as bulky groups were attached to base structure of furan and then docked to DNA there were less no of hydrogen bonding interactions; owing to more steric hindrances and consequently the fact that number of hydrogen bonding interactions decrease on attaching bulky groups complement the obtained results. These hydrogen bonding interactions were obtained from Discovery Studio Visualizer [31].

Figure 3: Best docked posed complexes for 4AH0 as obtained from UCSF Chimera.
| S. No. | Ligand | Binding Energy (kcal/mol) | Docking RMSD (Å) |
|---|---|---|---|
| 1 | Mol-1 | -12.03 | 6.165 |
| 2 | Mol-2 | -10.94 | 6.429 |
| 3 | Mol-3 | -12.06 | 6.163 |
| 4 | Mol-4 | -12.39 | 6.548 |
Table 2: Various docking results obtained for 4AH0DNA sequence.
Following Figures 4 and 5 represent the atomistic binding sites for each of the drug-DNA complexes and corresponding H-donor/ acceptor clouded regions at binding sites, due care while taking images regarding the same has been taken in order to have the same pose for both, binding site and H-donor/acceptor cloud, respectively. Further Table 3 represents the donor and the acceptor residues involved in the formation of hydrogen bond between the DNA and ligands.

Figure 4: H-bonds in best docked posed complexes for 4AH0 as obtained from Discovery Studio Visualizer.

Figure 5: H-bond donor and acceptor regions in best docked posed complexes for 4AH0 as obtained from Discovery Studio Visualizer.
| S. No. | Complex | No. of H-Bonds | Interacting Species | H-Bond Length (Å) |
|---|---|---|---|---|
| 1 | 4AH0+Mol-1 | 1 | H - DC11:O3' | 2.679714 |
| 2 | 4AH0+Mol-2 | 4 | H - DT8:O4' | 2.148102 |
| H - DT7:O2 | 3.06722 | |||
| H - DC11:O2 | 2.689916 | |||
| H - DC11:O3' | 2.256256 | |||
| 3 | 4AH0+Mol-3 | 2 | H - DC11:O4' | 2.153036 |
| H - DC11:O3' | 2.520974 | |||
| 4 | 4AH0+Mol-4 | 1 | H - DT9:O3' | 2.656984 |
Table 3: The donor and the acceptor amino acid residues involved in the formation of hydrogen bond between the DNA and ligand atoms.
Molecular dynamics simulation
Structural stability of biomolecules under dynamical conditions over a specifically mimicked environment for a pre-defined period of time can be studied via molecular dynamics simulations. Such studies hold significant importance in the structure and dynamics of biomolecules owing to less experimental costs. Various parameters were studied and analyzed for their contribution to stability analysis of the drug-DNA complexes, and are discussed as follows:
Variation in radius of gyration: In order to understand the dynamic stability and compactness of DNA-ligand complexes, Radii of Gyration values are determined. Clearly, the avg. radius of gyration lies between 1.25 nm to 1.50 nm resp. The collective Radius of Gyration variations for each drug-DNA pairs are shown in Figure 6. The Radius of Gyration data reveals that mol-2, mol-3 and mol-4 with 4AH0 sequence form themost stable complexes whereas for mol-1, the radius remains intact till ~3500ps but after that time limit there are frequent perturbations in the conformations of the complex owing to minimized stability.

Figure 6: Variations for Radius of Gyration.
Root mean square deviation: The RMSD is treated as the measure of the conformational stability of biological macromolecules. The plots for RMSD of all the drug- DNA complexes are represented in Figure 7. From the variations shown below, the RMSD for corresponding to mol-2, mol-3 and mol-4 with 4AH0 almost lie between 0 to 0.5 nm representing least or no deviations. However,4AH0+mol-1, shows deviations around 3400ps, 4000ps and 4500ps and hence confirms for having had formed least stable complex with considerable deviations in the DNA double helix.

Figure 7: RMSD for Drug-DNA Complexes.
Root mean square fluctuation: RMSF records the fluctuation of each amino acid base pair including the fluctuations in the flexible regions within the nucleic acid during the course of the MD simulation.Well-structured regions and loosely bound regions in DNA strands are distinguished by low and high root mean square fluctuations values respectively. For small proteins, a fluctuation between 1~3 Å is acceptable. The graphs shown below in Figure 8 suggest that, 4AH0+mol-2, 4AH0+mol3 and 4AH0+mol-4 had the least fluctuations, and especially around and beyond residue number 800 among and hence owe towards the stability of the complexes.Here this range lies between 0~0.5Å. Whereas,4AH0+mol-1 shows maximum deviations as seen from the corresponding graph.

Figure 8: RMSF for Drug-DNA Complexes.
Natural bond orbital analysis: Natural Bond Orbital (NBO) analysis played a crucial role for the calculation of intra and intermolecular orbital bonding and interaction in the optimized electronic chemical structure, particularly charge transfer and stabilization gain energy. Geometry, of the NBO analysis were optimized at the level of B3LYP/6-31G (d) method using Gaussian 09 program package (Figure 9).

Figure 9: Optimized geometries are showing the molecular orbital with maximum electron density.
The NBO analysis is a suitable source of the charge transfer donner to receptor in the orbital-based. The total hyper energy conjugated interaction (stabilization gained energy) from NBO donor to NBO acceptors were calculated by the second-order perturbative theory. The estimates of donor-acceptor interactions in the NBO basis and presented in the Table supporting information (SI) Table S1-S4. This analysis clearly indicates that the stabilization gained energy is a few times higher in the total stabilization gain energy [32].
Modeling techniques when applied in the studies of drugs and associated targets have always proven them to be key players in pharmacy industries. These techniques not only not only complement experiments but also help in the improvement of existing computational tools.
The current research article addressed a problem regarding interaction and stability of drugs in the vicinity of DNA, and our findings successfully resolve this claim. The optimized drugs structures when docked to selected DNA generated best docked pose by finding the most stable binding site, these docked complexes when subjected to molecular dynamics for time scale evaluation of their stability yielded similar results that complemented the docking results and previously reported datas. Our evaluated parameters viz, hydrogen bonding, Radius of Gyration, RMSD and RMSF were sufficient to support our claim. We successfully traced out the most stable complexes, 4AH0+mol-2, 4AH0+mol-3 and 4AH0+mol-4. This clearly raises numerous questions to be well addressed regarding their human bioassays, but we can say that in vivo experiments are solely responsible for any sure and certain conclusions. However, computational techniques have a very bright future in this field. This not only keeps the doors open for further research but also leaves us with strong and affirmative methodologies. The NBO analysis is provided the detailed insight into the type of hybridization and the nature of bonding in the particularly charge transfer and stabilization gain energy.
The authors declare no conflicts in their mutual interests, in any manner whatsoever.
Anwesh Pandey acknowledges to the University Grants Commission, India for financial assistance during the course of this research work and Suresh Kumar is also grateful to the University Grants Commission for financial support through the Dr. D. S. Kothari Postdoctoral Fellowship (No.F.4-2/2006 (BSR)/PH/18-19/0022).
Citation: Pandey A, Upadhyaya A, Kumar S, Yadav AK (2020) Interaction, Dynamics and Stability Analysis of Some Minor Groove Binders With B-DNA Dodecamer 5’-(CGCAAATTTGCG )-3’. Drug Des. 10:172.
Received: 11-Nov-2020 Accepted: 25-Nov-2020 Published: 02-Dec-2020 , DOI: 10.35248/2169-0138.21.10.172
Copyright: © 2020 Pandey A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.