Pancreatic Disorders & Therapy
Open Access
ISSN: 2165-7092
ISSN: 2165-7092
Review Article - (2015) Volume 0, Issue 0
Sleep apnea syndrome (SAS) a highly prevalent disorder, and characterized by repetitive episodes of intermittent hypoxia (IH), i.e. recurrent episodes of oxygen desaturation during sleep, the development of daytime sleepiness, and deterioration in the quality of life. Multiple epidemiological studies have provided evidence implicating the presence of SAS as a risk factor for insulin resistance and type 2 diabetes and have reported that type 2 diabetes is associated with SAS independently of age, sex, and body habitus. One of the postulated mechanisms for the metabolic alterations associated with SAS is that IH leads to substantial alterations in both pancreatic β cell function and organ glucose homeostasis. On the other hand, hyperglycemia is known to increase the rate of β cell replication, which can provide an increased source of insulin to combat insulin resistance. Although accumulating evidence suggests associations between SAS and type 2 diabetes, the direct effect of IH on pancreatic β cell has been unknown. In this review, we focus on the impact of IH on pancreatic β cells, particularly β cell dysfunction and cell proliferation.
<Keywords: Type 2 diabetes, Intermittent hypoxia, Glucose-induced insulin secretion, Pancreatic β cell proliferation, Regenerating gene, Hepatocyte growth factor, Sleep apnea syndrome
Sleep apnea syndrome (SAS) is characterized by recurrent episodes of oxygen desaturation during sleep, the development of daytime sleepiness, and deterioration in the quality of life [1], and this condition affects up to 32% of the adult population [2]. Numerous epidemiological and clinical studies [3] have confirmed that SAS is closely related to type 2 diabetes and that this relationship is independent of confounding factors, such as obesity and family history. During sleep, the repeated upper airway collapse in SAS patients can cause serious recurrent apnea, leading to the characteristic intermittent hypoxia (IH). Thus, patients are exposed to an alternation of low oxygen tension and normal oxygen tension. IH is similar to the oxygen abnormality in the ischemic/reperfusion process and can initiate oxidative stress. At present, SAS is considered an oxidative stress-induced disease [4].
Obesity is recognized as a major health care problem, and there is a high prevalence in the association with metabolic syndrome, the commonly used term for the cluster of obesity, insulin resistance, hypertension, and dyslipidemia [5,6]. Obesity is a predominant risk factor for SAS [7], insulin resistance, hyperglycemia, and type 2 diabetes [8]. Multiple epidemiological studies have provided evidence implicating the presence of SAS as a risk factor for insulin resistance and type 2 diabetes [9], the latter of which was reported to be associated with SAS independently of age, sex, and body habitus [10]. It has also been reported that the prevalence of type 2 diabetes increases with the severity of SAS and that the severity of SAS is associated with poor diabetic control in patients with type 2 diabetes [11]. The association between nocturnal IH and the risk of developing type 2 diabetes among community-dwelling Japanese participants was also reported [12].
One of the postulated mechanisms for the metabolic alterations associated with SAS is that IH during sleep, a hallmark of SAS, leads to substantial alterations in both pancreatic β cell function and organ glucose homeostasis. The progression to type 2 diabetes depends on both the impairment of glucose-induced insulin secretion from pancreatic β cells and the presence of insulin resistance. On the other hand, hyperglycemia is known to increase the rate of β cell replication [13, 14], which can provide an increased source of insulin to combat insulin resistance. In a mouse model, IH was found to cause β cell replication without hyperglycemia [15,16], suggesting a possible mechanism that IH directly stimulates β cell replication. Here, we focus on the impact of IH on pancreatic β cells, particularly β cell dysfunction and proliferation, using an in vitro IH system [17,18].
We exposed hamster HIT-T15 β cells, established by transforming Syrian hamster (Mesocricetus auratus) primary culture cells with SV40, contain modest numbers of membrane-bound secretory granules and have the ability to release insulin following glucose stimulation, similar to that of isolated pancreatic islets [19], to normoxia, IH, or sustained hypoxia for 24 hours (Figure 1) and found that glucose-induced insulin secretion from IH-treated HIT-T15 β cells was significantly attenuated. The glucose-induced insulin secretion was also attenuated in isolated rat islets by the treatment of IH.
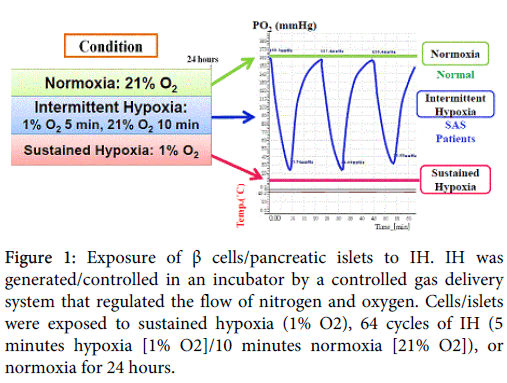
Figure 1: Exposure of β cells/pancreatic islets to IH. IH was generated/controlled in an incubator by a controlled gas delivery system that regulated the flow of nitrogen and oxygen. Cells/islets were exposed to sustained hypoxia (1% O2), 64 cycles of IH (5 minutes hypoxia [1% O2]/10 minutes normoxia [21% O2]), or normoxia for 24 hours.
On the other hand, the levels of insulin mRNAs in HIT-T15 cells (hamster insulin mRNA) and rat islets (rat Ins1 and Ins2 mRNAs) were unchanged by IH treatment, suggesting that IH attenuates glucose-induced insulin secretion without changing insulin gene transcription [17]. Recent reports of β cell dysfunction in vivo IH exposure animal models [20-22] also support the concept of pancreatic β cell dysfunction including glucose-induced insulin secretion by IH exposure.
We then examined the mRNA levels of several genes involved in glucose-induced insulin secretion and found that the mRNA levels of glucose transporter 2, glucokinase, sulfonylurea receptor 1, and the α 1c subunit of voltage-dependent L-type calcium channels were unchanged between IH-treated and normoxia-treated islets [17]. These results suggest that IH has no effect on the gene expression concerning Ca2+ influx from extracellular sources for glucose-induced insulin secretion.
We then analyzed genes involved in Ca2+ mobilization from intracellular pools, such as CD38, type 2 ryanodine receptor Ca2+ channel, and FK506-binding protein 12.6 and found that the mRNA level of CD38 was significantly lower in IH-treated islets than in normoxia-treated islets [17]. To investigate whether IH inhibits CD38 transcription, HIT-T15 cells were transiently transfected with the reporter plasmid consisting of a luciferase reporter gene under the control of human CD38 promoter. After exposure to IH for 24 hours, we measured luciferase activities of normoxia- and IH-treated cells and found that the transcriptional activity of CD38 was attenuated by IH [17].
Cyclic ADP-ribose (N1-(β-D-ribosyl)adenosine 5'(P1),5''(P2)-cyclic diphosphate: cADPR) acts as a second messenger for Ca2+ mobilization from an intracellular Ca2+ pool for glucose-induced insulin secretion [23-26], and CD38 has both ADP-ribosyl cyclase and cADPR hydrolase (EC 3.2.2.6) activities for cADPR synthesis from NAD and hydrolysis to ADP-ribose (adenosine 5'-(5-deoxy-D-ribofuranos-5-yl diphosphate)), respectively [25-27]. CD38 was reported to play an essential role in glucose-induced insulin secretion [24-28]; down-regulation of CD38 mRNA was reported in the islets of Goto-Kakizaki (GK) rat [29], a rodent model of spontaneously occurring type 2 diabetes with impaired glucose-stimulated insulin secretion. In addition, a single nucleotide polymorphism that decreases the cADPR synthesizing activity of CD38 [30] and autoantibodies against CD38 that attenuated cADPR synthesis [31,32] were detected in type 2 diabetes patients. We found that IH suppressed CD38 transcription as well as glucose-induced insulin secretion. Therefore, it is possible that the IH-induced attenuation of glucose-induced insulin secretion is mediated by the suppression of CD38 expression at transcriptional level in β cells. To verify this possibility, we prepared CD38 expression vector under the control of cytomegalovirus promoter and transfected it into HIT-T15 β cells. After the exposure to normoxia or IH for 24 hours, HIT-T15 cells transfected with CD38 expression vector showed increased insulin secretion by glucose stimulation whereas the glucose-induced insulin secretion from HIT-T15 cells transfected with control vector did not [17], suggesting that the suppression of CD38 expression in IH-treated β cells is crucial for the attenuation of glucose-induced insulin secretion. Recently, Wang et al. also reported that glucose-induced insulin secretion but not KCl-induced insulin secretion was impaired in IH-treated mice [33]. As CD38 knockout mouse islets showed similar insulin secretion properties with those of IH-treated mouse islets (impairment of glucose-induced insulin secretion but unaffected in KCl-induced insulin secretion) [28], our results obtained in vitro experiments possibly occur in SAS patients and may make them diabetes.
In addition to glucose-induced insulin secretion, we have recently showed that IH stimulation increased selenoprotein P mRNA but no other diabetes-associated hepatokine (fibroblast growth factor 21, α2-HS glycoprotein, angiopoietin-like protein 6, horomone-binding globulin, leukocyte cell-derived chemotaxin 2, and lipasin) mRNAs in cultured hepatocytes and that the concentrations of selenoprotein P, determined by ELISA in the cultured hepatocyte medium, were significantly increased in the IH-treated hepatocytes [34]. These results strongly suggest that cyclic changes of hypoxia-reoxygenation, which occurs in SAS patients, up-regulate selenoprotein P, accelerating insulin resistance in SAS patients to exacerbate their diabetes.
To evaluate the direct effects of IH on β cell proliferation, rat RINm5F (derived from New England Deaconess Hospital rat insulinoma induced by high-dose X-ray irradiation), hamster HIT-T15, and human 1.1B4 β (generated by electrofusion of normal human islet cells with immortal human PANC-1 pancreatic duct cell line [35]) cells were exposed to normoxia, IH, or sustained hypoxia for 24 hours. The viable cell numbers of RINm5F, HIT-T15, and 1.1B4 β cells determined by WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)- 5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] cleavage method were significantly increased by IH, whereas the cell numbers decreased in RINm5F and HIT-T15 cells and were unchanged in 1.1B4 cells by sustained hypoxia. To determine whether IH decreases apoptosis, we next measured apoptosis of RINm5F and HIT-T15 cells by TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) method, and found no statistically significant difference in apoptosis between normoxia- and IH-treated β cells. These results indicate that IH directly increases β cell proliferation [18]. The results were well fitted the results that pancreatic β cell proliferation in vivo mouse model of intermittent hypoxia [15].
Regenerating gene (Reg) family genes, which encode autocrine/paracrine β cell growth factors, are reported to be involved in β cell proliferation [25,36-40]. We analyzed the mRNA levels of Reg family genes by real-time RT-PCR and found that the mRNA levels of Reg I, PAP II/Reg III, PAP III, and Reg IV significantly increased in rat RINm5F β cells by IH stimulation. The mRNA level of REG Iα was significantly increased in human 1.1B4 β cells by IH [18]. In order to evaluate independent Reg family members, we introduced small interfering RNAs (siRNAs) for rat Reg mRNAs into RINm5F β cells and measured IH-induced cell proliferation by WST-8 assay. We found that the introduction of siRNAs for Reg I, PAP II/Reg III, PAP III, and Reg IV suppressed IH-induced β cell proliferation, whereas siRNA for PAP I/Reg2 did not change the cell numbers [18], indicating the involvement of Reg family genes, at least Reg I, PAP II/Reg III, PAP III, and Reg IV in rat, in IH-induced pancreatic β cell proliferation.
In contrast to pancreatic β cells, IH stimulation increased hepatocarcinoma-intestine-pancreas (HIP)/pancreatitis-associated protein (PAP) mRNA but not other members of the REG family gens in human hepatocytes [34]. As HIP/PAP was originally found as a gene expressed in hepatocarcinomas [41] and recent transgenic/knockout mouse experiments showed its mitotic/growth factor activity to hepatocytes and its obesogenic properties [42-44], over-expression of HIP/PAP, induced by IH, could induce proliferation of hepatocytes and adipocytes [44]. The hepatocytes exposed to IH are also induced to express selenoprotein P [34]. Thus, IH treatment could induce selenoprotein P-expressing hepatocyte proliferation as well as pancreatic β cell proliferation.
As Reg family genes are activated by IL-6 [25,40,45-51], we tested whether IL-6 also increases human REG Iα mRNA in human pancreatic β cells and found that IL-6 significantly increased human REG Iα mRNA in 1.1B4 human β cells. We then determined IL-6 mRNA expression in RINm5F and 1.1B4 β cells exposed to IH and found that the mRNA levels of IL-6 in RINm5F and 1.1B4 cells were significantly increased by IH [18], suggesting that IH stimulation up-regulates Reg family mRNAs through IL-6 expression in pancreatic β cells. Recently, up-regulation of IL-6 in pancreatic islets of in vivo mouse IH model was also reported [21]. As IL-6 has been also reported to down-regulate its signaling molecules, such as IL-6 receptor and gp130, we measured mRNA levels of IL-6 receptor and gp130 in RINm5F cells, rat islets, and 1.1B4 cells exposed to normoxia, IH, or sustained hypoxia. We found that no IH-treated cells (RINm5F cells, rat islets, and 1.1B4 cells) showed down-regulation of IL-6 receptor/gp130 mRNAs [18], suggesting that IL-6, induced by IH, up-regulates Reg family mRNAs to stimulate β cell proliferation.
Although the Reg protein is a pancreatic β cell growth factor, a high concentration of the reportedly induces β cell apoptosis [18,52,53]. These findings suggest that IH-stimulated β cells express some anti-apoptotic factors against a high concentration of Reg protein. HGF has been shown to protect rat RINm5F β cells against free fatty acid-induced apoptosis [54]. Therefore, we tested a possible anti-apoptotic action of HGF against a high concentration of Reg protein (1000 nM) and found that HGF (both 2.5 and 25 ng/ml) protected RINm5F β cells against apoptosis induced by a high concentration of rat Reg I protein [18,53]. We next examined whether HGF expression is induced by IH and found that HGF mRNA was induced by IH in RINm5F and 1.1B4 β cells [18]. HGF gene was reported to be induced by IL-6 stimulation via STAT3 activation [53] and IL-6 was up-regulated in IH-treated pancreatic β cells in vitro and in vivo [18,21]. Therefore, IH stimulation induces HGF gene expression via the IL-6/STAT3 pathway in pancreatic β cells. As HGF inhibited high-concentrations of Reg I (a member of Reg family protein)-induced β cell apoptosis [18,53], this simultaneous gene expression of HGF with Reg family genes may work as a safety valve for pancreatic β cells to minimize apoptotic effects of Reg family proteins.
A possible link between SAS and cancer has recently been suggested [55,56] and possible involvements of vascular endothelial growth factor, hypoxia-inducible factor, and reactive oxygen species-activated activator protein 1 and nuclear factor-κB in cancer development/progression in SAS patients were reported [57]. Recently, enhanced metastatic ability of IH-treated human pancreatic cancer cell lines (PANC-1 and BxPC-3) was also reported [58]. As Reg family proteins and HGF were reported to be over-expressed in some cancer cells and to influence cancer patient prognosis [41,59-72], IH may accelerate tumor proliferation and/or invasion, ultimately resulting in an adverse outcome via over-expression of Reg family and/or HGF.
Recently, there has been great interest in the interaction between SAS and metabolic dysfunction. SAS is commonly found in patients with type 2 diabetes. In addition to the development of glucose intolerance and insulin resistance, the progression to type 2 diabetes is dependent on the impairment of glucose-induced insulin secretion from pancreatic β cells and the compensatory replication of pancreatic β cells to combat the presence of insulin resistance. However, the direct effects of SAS/IH on β cells have not been clarified. We examined the relationship between IH and β cell dysfunction and/or replication of pancreatic β cells and their mechanisms.
Our studies indicated that that the cyclic change of hypoxia-reoxygenation, which occurs in SAS patients, attenuates glucose-induced insulin secretion from pancreatic β cells via CD38 down-regulation, resulting in glucose intolerance and type 2 diabetes, as shown in Figure 2. IH also increased β cell mass by up-regulation of Reg and HGF to overcome reduced glucose-induced insulin secretion and insulin resistance, causing hyperinsulinemia and making patients more obese; this worsened their SAS anatomically. The feature of gene expression in IH-treated pancreatic β cells was in agreement with that found in GK rat islets which showed down-regulation of CD38 [29] as well as up-regulation of Reg family genes [73].
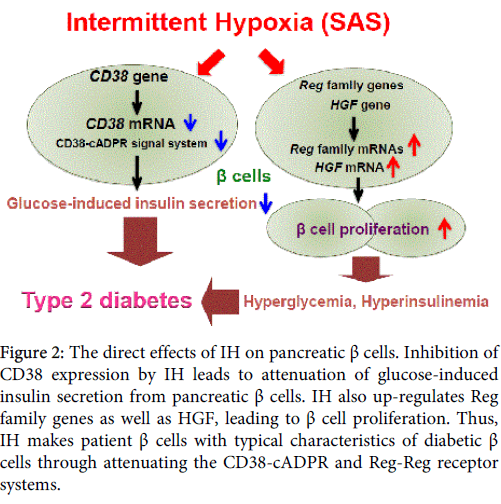
Figure 2: The direct effects of IH on pancreatic β cells. Inhibition of CD38 expression by IH leads to attenuation of glucose-induced insulin secretion from pancreatic β cells. IH also up-regulates Reg family genes as well as HGF, leading to β cell proliferation. Thus, IH makes patient β cells with typical characteristics of diabetic β cells through attenuating the CD38-cADPR and Reg-Reg receptor systems.
The authors declare that there are no conflicts of interest.
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (Grant nos. 23591158, 23659161, and 15K19425) and the Japan Science and Technology Agency. We are grateful to Drs. A. Itaya-Hironaka, A. Yamauchi, Y. Kyotani, R. Shobatake, and Y. Dohi (Nara Medical University) for their help and useful suggestions.