Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Research Article - (2018) Volume 6, Issue 2
Background: Microscopic parasite detection during the course of treatment or follow-up suggests that a proportion of ACT treated children in Kenya do not completely clear Plasmodium falciparum parasitemia. Plasmodium falciparum mutation in chloroquine resistance transporter gene (Pfcrt76), multidrug resistance gene1 (Pfmdr1), deubiquitinating enzyme gene (Pfcubp-1) and clathrin vesicle associated adapter 2, u subunit encoding gene (Pfap2mu) and multidrug resistant protein 1 gene (Pfmrp1) have been associated with subsequent patent recrudescence after ACT treatment. As there are no validated markers of ACT resistance in Africa to-date, surveillance of changes in these polymorphisms during the course of treatment is useful in establishing their role in ACT treatment outcome. Methods: 118 P. falciparum 2013 to 2015 samples from ACT clinical efficacy studies were genotyped for frequency of drug resistance polymorphisms using sequence analyzers. Each sample was screened at least three to four-time points namely; day zero before start of treatment then days 2 and 3 after initiation of treatment plus the day of subsequent parasitemia by microscopy prior to day 42 for some of the subjects. Sequence analyzers were used to genotype for frequency of drug resistance polymorphisms, Pfmdr1 gene copy number and genetic diversity typing of the 12 microsatellite loci. Genetic diversity of parasite populations across four time-points was determined by analysis of 12 microsatellite loci. Worldwide Antimalarial Resistance Network’s parasite clearance estimator (PCE) was used to determine parasite clearance rates. Results: The new genes Pfap2mu and Pfubp had S145C and E1528D being most polymorphic with prevalence’s of 18% and 19%, respectively Pfmdr1 86,184 and 1246 had significant increase in wild type alleles between day zero and time-points 3 and 4. Microsatellite profile analysis show that the mean number of alleles in all the loci across the 8 populations ranged from 9.250 to 1.000. Poly α was the most polymorphic with 35 alleles. The mean unbiased HE was 0.672 while Shannon diversity index for the 8 populations was ranging between 0.182-0.000, none of the parasite analyzed had matching haplotypes. The mean parasite clearance half-life was 2.63 h (95% confidence interval [CI]) and the median clearance half-life was 2.24 h. The parasite clearance half-life ranged from 1.14-5.05 h. Conclusion: Increased wild-type Pfmdr1 86,184 and 1246 as well as polymorphisms in Pfap2mu and Pfcubp-1 in post day zero suggest that these genes could be responding to ACT dosing and therefore require continued monitoring. Samples with multiple copies of the Pfmdr1 did not indicate show effect on parasite clearance rate. Though there was no correlation between these profiles and clearance rates, further evaluations are needed to determine the potential public health implications of these observations and the utility of these loci as markers of artemisinin sensitivity in populations of P. falciparum worldwide.
Keywords: Microscopic parasite detection; Artemisinin resistance; Plasmodium falciparum; Mutation; Genetic diversity; Antimalarial resistance; Antimalarial
Artemisinin-based combination therapy (ACT) was recommended by the WHO in 2001 for treatment of uncomplicated Plasmodium falciparum malaria following development of resistance to sulphadoxine- pyrimethamine (SP) [1]. The successes of this treatment is dependent on the rapidly acting artemisinin derivatives which have a short half-life as the key drug to reduce the parasite biomass rapidly alleviating symptoms and the partner drug with a relatively longer half-life to clear the residual parasites [2]. However, multiple studies signal a trend of reversal of these gains by reducing efficacy of ACT. Previous studies showed that parasites in Cambodia and its surroundings have slow parasite clearance among patients under artesunate mono therapy use [1,3] and recently against ACTs [4]. This is rather disturbing, first, because malaria parasites have developed resistance to all the other classes of antimalarials that could serve as alternative treatments. Secondly, there is abject lack of new antimalarial molecules in late stage product development that can be deployed within the next 5 years for malaria treatment. These factors emphasize the need for accurate characterization of ACT resistance globally to allow innovation of strategies of deployment of these treatments, suitable for prolongation of their lifespan in order to sustain reduction in morbidity and mortality.
Resistance to ACTs in Southeast Asia (SEA) characterized by slow clearance rates of >6.6 h, mutations in Kelch propeller gene [5,6] and increases Increased Tolerance to Artemisinin by 0-3 h old ringstage parasites [7]. Analyses of parasites from Africa show absence of these mutations [8,9]. However, mutation in chloroquine resistance transporter gene (Pfcrt76), multi drug resistance gene1 (Pfmdr1), deubiquitinating enzyme gene (Pfubp1) and clathrin vesicle associated adapter 2, u subunit encoding gene (Pfap2mu)) have been associated with non-responsiveness to artemisinins in parts of Kenya [2,10,11]. If verified, these markers would be useful in detecting emerging resistance to ACTs in other regions outside SEA. This study characterized strain and polymorphisms content of declining infections of 118 subjects during ACT treatment in clinical efficacy studies and compared with treatment outcomes including parasite clearance rates.
One hundred and eighteen individuals aged six months and over presenting at Kombewa sub-county hospital with uncomplicated falciparum infection were enrolled in an artemisinin combination therapy efficacy study after signing informed consents/assents. The study was approved by the Ethical Review Committee of the Kenya Medical Research Institute (KEMRI), Nairobi, Kenya and Walter Reed Army Institute of Research (WRAIR) Institutional Review Board, Silver Spring, MD under protocols KEMRI-SSC 2518/WRAIR 1935a and WRAIR 1935b. Protocols used in these studies complied with International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines. These studies were conducted in accordance with the Declaration of Helsinki and the Belmont Report including all federal regulations regarding the protection of human participants as described in 32 CFR 219 (The Common Rule). Internal policies for human subject protections and the standards for the responsible conduct of research of the US Army Medical Research and Materiel Command were also followed. WRAIR holds a FederalWide Assurance from the Office of Human Research Protections under the Department of Health and Human Services. Accordingly, all key study personnel in both studies were certified as having completed mandatory human research ethics education curricula and training under the direction of the WRAIR IRB Human Subjects Protection Program.
Each individual was monitored for parasite clearance by microscopy during the 3 days hospitalization period and subsequent parasitemia on day 7, 14, 21, 28, 35 and 42 in accordance with WHO standard clinical efficacy study guidelines. For the purpose of this study, the samples were further diagnosed by PCR for submicroscopic parasitemia. Plasmodium falciparum positive samples at three to four time-points namely time-point zero before administration of the first dose of ACTs followed by days 2 and 3 after initiation of treatment and on the day of treatment subsequent parasitemia during the 42 days follow up period were collected.
Genomic DNA was extracted from 200 μl of day zero whole blood samples and FTA dry blood spots for subsequent time point’s samples as described by the Qiagen kit manufacturer (QIAmp DNA Mini handbook 04/2010). Microsatellite genotyping was done using a heminested PCR strategy to amplify twelve microsatellite loci distributed across the P. falciparum genome as previously described. The loci assayed include: Poly ᾳ (Chr4), TA42 (Chr5), TA81 (Chr5), TA1 (Chr6), TA109 (Chr6), TA87 (6), TA40 (Chr10), 2490 (Chr10), ARAII (Chr11), PFG377 (Chr12), PFPK2 (Chr12) and TA60 (Chr13). Four loci (Poly ᾳ, ARAII, pfG377and PfPk2) are in coding sequences from GenBank.
Fragment analysis on PCR products by capillary electrophoresis (CE)
Sequencing of purified PCR product in both directions was then performed using ABI PRISM BigDye terminator V3.1 sequencing kit as specified by the manufacturer (Applied Biosystems CA; USA). The BigDye PCR reaction was performed in 15 μl for each primer with 5 μl of DNA template and 1 μl of 4 μM primer. Reaction conditions consisted of an initial denaturation at 95°C for 5 min followed by 35 cycles of 95°C for 15 s, 55°C for 30 s and 68°C for 2 min 30 s and a final extension step for 3 min at 68°C. The chain terminated products were cleaned using a sephadex column by spinning at 2130 rpm for 5 min to ensure only pure sequenced products were eluted. The pure product was then hydrated with 10 μl of HIDI formamide (Applied biosystems) The samples were then denatured at 95°C for 5 min and chilled in ice to keep the DNA fragments single stranded prior to loading on the the 3130x series genetic analyzer (Applied Biosystems).
The CE output was then exported to GeneMapper software version 4.0 (Applied Biosystems) for fragment analysis which generates estimates of DNA fragments relative to the size standard. DNA fragment size calculated in base pairs and peak heights, calculated in Relative Fluorescence Units - RFU were obtained. GeneMapper software v4.0/4.1 uses the parameters to quantify samples containing multiple alleles per locus.
Detection of genetic polymorphisms and copy number estimation
Genomic DNA for day 0, days 2, 3, 7, 14, 28and 42 was amplified at selected loci and polymorphisms in the Pfcrt K76T, Pfmdr1 N86Y, Y184F and D1246Y alleles determined by sequencing or PCRbased single-base extension on Sequenom MassARRAY platform (Agena Biosciences, San Diego, CA, USA) following manufacturer recommendations. Previously published primers for Pfcrt K76T and Pfmdr1 SNPs for allele polymorphisms were used for PCR and sequencing analyses. Concentration of PCR reactants, purifying of the successfully amplified targets for eventual sequencing or PCR-based single-base extension assays followed by contig assembly ad analysis were done as previously described [12-14]. Polymorphisms in the Pfap2mu and Pfubp1genes were determined by direct sequencing of 3 fragments encompassing codons 1-174, 121-399 and 377-621 in the former and codons 1463–1563 for the later as described by Henriques et al. [2].
Estimation of Pfmdr1 copy number
Pfmdr1 gene copy number was estimated as previously described using the ABI 7500 real-time fast qPCR system (Applied Biosystems Inc., Foster City, CA). Each isolate was assayed in triplicate and analyzed with SDS software (version 2.0.6; Applied Biosystems Inc., Foster city, CA). Reference strains 3D7 which carries one Pfmdr1 gene and Dd2 which carries 2-3 Pfmdr1 gene copies were used as copy number controls in each run. The reactants comprised 10 μM working stock of primers (10 μl of 100 μM stock in 90 μl of dH2O) and 10 μM working stock of probe (10 μl of 100 μM stock in 90 μl of dH2O) is reconstituted and The assay conditions were; 1 cycle at 50°C for 1 min, 1 cycle at 95°C for 10 min and 2-step PCR 95°C for 15 s and 58°C for 1 min for 45 cycles. The relative quantification technique 2-^^CT was used to estimate the copy number variation. Samples with less than 1.75 were considered to have true Pfmdr1 gene amplification.
Parasite clearance rate calculations
Parasitemia was estimated every 6 h by examining Giemsa-stained thin and thick blood smears in 200 high power fields. Parasite density per micro liter was estimated from the number of parasites counted per 2000 WBCs and based on the total number of WBCs from the complete blood count. Parasite clearance rates were calculated using the Worldwide Antimalarial Resistance Network (WWARN) tool for Parasite Clearance Estimator (PCE) located at http://www.wwarn. org/toolkit/data-management/parasite-clearance-estimator. Log transformed parasite density was plotted against time in hours to generate the slope half-life which is defined as the time needed for parasitemia to be reduced by half. This constant is independent of the starting value of parasitemia [15]. The half-life was calculated as follows:
T1/2=loge (2)/k
T=0.692/k
Where, k is the clearance rate constant
The study is a retrospective study, under a two-arm randomized open-label trial assessing the therapeutic efficacy of artemisinin-based combination therapies for the treatment of uncomplicated malaria in Kisumu County, Western Kenya.
Clearance slope half-life
Parasitemia decreased gradually and fully resolved by end of 72 h follow-up period among all cases. Analyses of the clearance rates by the Worldwide Antimalarial Resistance Network’s Parasite clearance estimator showed a median of 4.00 h (interquartile range (IQR), 4.00- 11.00, 95% confidence interval (CI)). The parasite clearance slope halflife depicted as the time needed for parasitemia to be reduced by half against the fraction normalized by which parasite count falls per unit time was in the range of 4.00 to 11.00 h (Tables 1a and 1b and Figure 1).
| Statistic Number of profiles with lag phase |
45 |
|---|---|
| Duration of lag phase (hours) Median | 4.00 |
| Range* | (4.00, 11.00) |
| IQR** | (4.00, 4.00) |
Table 1a: Summary of individual patient lag phase (hours).
| Statistics | |||
|---|---|---|---|
| Median | Range* | IQR** | |
| PC50 | 2.21 | (0.24, 11.57) | (3.62, 7.67) |
| PC90 | 8.76 | (3.17, 18.26) | (8.74, 13.61) |
| PC95 | 11.58 | (4.42, 21.54) | (10.87, 15.73) |
| PC99 | 18.13 | (7.32, 30.48) | (15.40, 20.84) |
Table 1b: Summary of time (hours) to clear 50%, 90%, 95% and 99% of parasitemia.
)
*Range=(minimum, maximum
**IQR: Inter Quartile Range (25th centile, 75th centile).
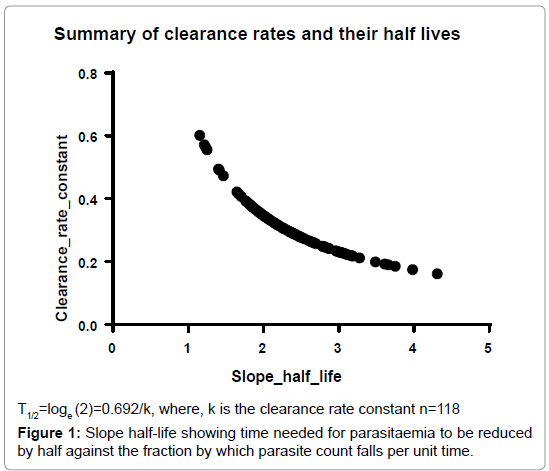
Figure 1: Slope half-life showing time needed for parasitaemia to be reduced by half against the fraction by which parasite count falls per unit time.
Copy number variations
The median copy number for n initial (day 0) samples was 1.01, IQR 0.5-1.2, 95%CI (Figure 2). The median copy no for subsequent timepoints was 1.01 day 1, 1.00 day 2 and 1.00 for subsequent parasitemia. This similarity in copy number median across time points suggests lack of involvement of the current treatment in copy number-driven selection (Figure 2).
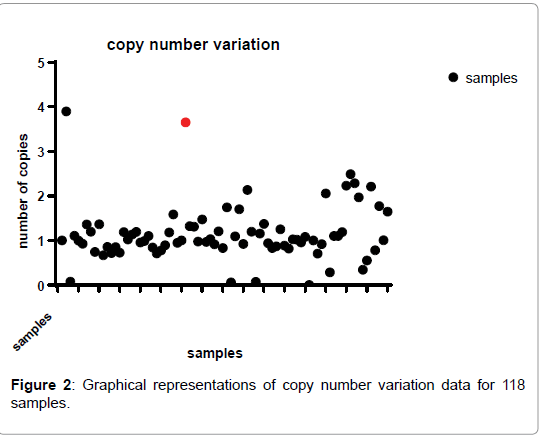
Figure 2: Graphical representations of copy number variation data for 118 samples.
P. falciparum multi drug resistant gene 1 (Pfmdr1) and p. Falciparum chloroquine resistant transporter gene (Pfcrt)
Frequencies of polymorphisms of three loci comprising Pfmdr1 86, 184 and 1246 alongside Pfcrt 76, that are known to confer resistance to antimalarials are summarized in Figure 3 and 4.117 sequences of Pfmdr1 gene that were successfully analyzed revealed three haplotypes based on the polymoprhisms at loci N86Y, Y184F and D1246Y. The most prevalent haplotype of Pfmdr1 among day zero samples was NFD at codons 86 and 1246 with 55%, present in at least 80% pre-treatment isolates exclusive of follow-up samples followed by NYD and NFY with 4% (codons 184 and 1246). The wild type locus Y184 was confirmed in 80% (n) of the 117 evaluable days zero samples, 48% (n) of which contained mutant genotype 184F. The remaining 19.26% (n) samples were mutant 184F. The prevalence of these NFD haplotype increased three-fold from 19% (n=25) in initial samples to 58% in the subsequent time-points suggesting selection during dosing with a p-value 0.001 and a, 95% CI.
All the 116 samples successfully typed for chloroquine resistant transporter gene (Pfcrt) were wild type (Figure 3).
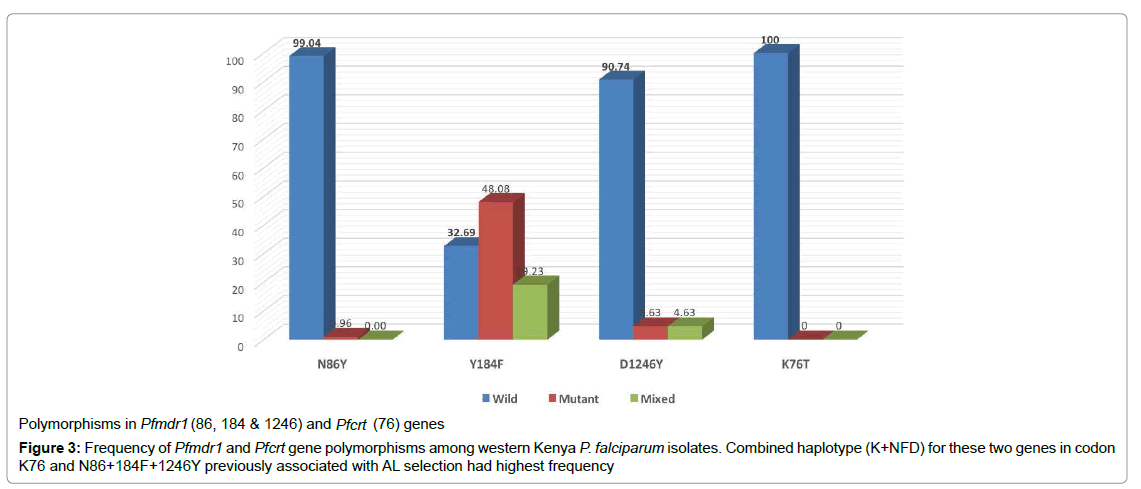
Figure 3: Frequency of Pfmdr1 and Pfcrt gene polymorphisms among western Kenya P. falciparum isolates. Combined haplotype (K+NFD) for these two genes in codon K76 and N86+184F+1246Y previously associated with AL selection had highest frequency.
Plasmodium falciparum clathrin vesicle associated adapter 2 (Pfap2mu)
The 2556 base pair-long Plasmodium falciparum Clathrin vesicle associated adapter 2 (Pfap2mu) was divided into three fragments with primers overlapping to cover the fragment for ease of sequencing. Fragment 1 covered 578 bp from position 53 outside the gene to 360 within the gene fragment 2 had 841 bp from 360-1199 while fragment 3 entailed 751 bp at position 1127 to 2556 inclusive of the flanking regions. Analysis of polymorphisms within this fragments showed that fragment 1 was the most polymorphic with 26 SNPS being detected within this fragment1 at frequencies of less than 20%. Of the 26 SNPS detected, S145C had the highest frequency 18% followed by L29F while N57Y, K25Q, T24S, I16 L, I10V with less than 1% were the least frequent. Polymorphism S145C that was lowest in initial samples rose to 18% in the subsequent parasitemia P=0.002 and 95% CI suggesting that it is involved in selection.
Fragment two had the following mutations A188G, A200T, T233G having frequencies of 10, 5 and 2%, respectively. Of the three mutations identified none of them was directly associated with clearance rates. One locus I112 L/M had bases alternating at a 0.5% frequency (Figure 4).
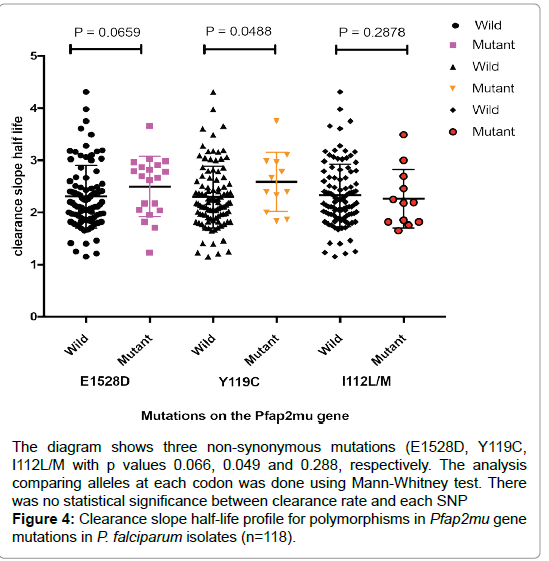
Figure 4: Clearance slope half-life profile for polymorphisms in Pfap2mu gene mutations in P. falciparum isolates (n=118).
Plasmodium falciparum deubiquinating enzyme gene (Pfubp1)
An optimized amplification strategy was designed for a region of Pfubp1 from codons 1463-1563, encompassing the polymorphic codon 1528 associated with in vitro parasite responses to artemisinin in Kenya [11]. Successful direct sequencing of this region in 118 pretreatment isolates identified haplotypes defined by 2 synonymous and 20 non- synonymous point mutations. 13 SNPS were detected within the Pfubp1 gene at frequency 18% for E1528D as the highest followed by K1535E and N1540H each having 10% while D1557N, E1578K and N1582D had less than 1%. The mutations were not directly associated with clearance rates (Figure 5).
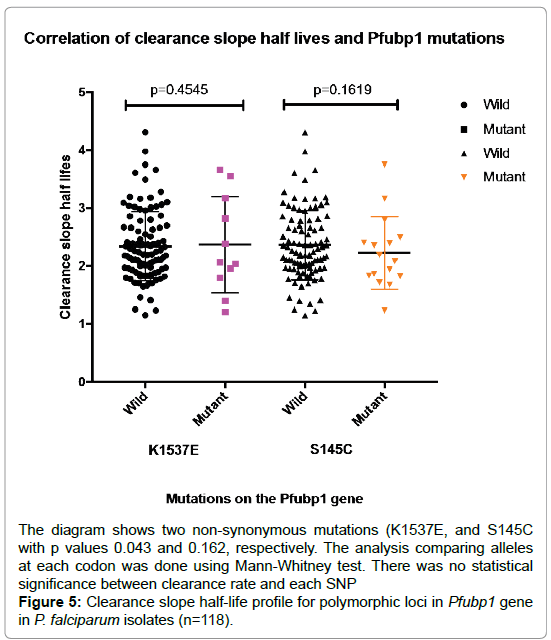
Figure 5: Clearance slope half-life profile for polymorphic loci in Pfubp1 gene in P. falciparum isolates (n=118).
Parasites population structure
To better understand origin of variability in clearance rates, a subset of samples that showed positive parasitemia beyond 24 h time point were analyzed for parasite clone composition. These samples obtained at time-points hour 0, 24 and hour 30 were 118; hour 36 had 80 samples, hour 42 had 40, hour 48 had 20 samples while D28 D35 and D42 had 14, 10 and 8, respectively. Samples at each of the nine time-points have survived same selection pressure hence referred to as population 1 to 9 for structure analyses. The 12 MS were highly polymorphic showing highly diverse parasites. The mean number of alleles in all the loci across the 8 populations ranged from 9.250-1.000 Polyα was the most polymorphic with 35 alleles whereas ARA2 was the least polymorphic locus with 5 alleles. From the number of distinct alleles observed at each locus, the unbiased HE estimated per locus is summarized in Table 2a. The mean unbiased HE was 0.672. Alleles were distributed widely across the populations where population 1 and 2 had a large number of private alleles of 1.750 compared to the other populations. The Shannon diversity index for the 8 populations was ranging between 0.182-0.000, further confirming higher genetic diversity in these populations (Tables 2b and 3 and Figure 6).
| Prevalence Day n= | % sensitivity |
|---|---|
| Hour 0, n=118 | 97% |
| Hour 24, n=118 | 47 |
| Hour 30, n=118 | 15 |
| Hour 36, n=80 | 10 |
| Hour 42, n=40 | 12 |
| Hour 48, n=20 | No data |
| Day 28, n=14 | 4% |
| Day 35, n=10 | No data |
| Day 42, n=8 | No data |
Table 2a: Unbiased HE estimated per locus.
| Mean values | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Population | Pop1 | Pop2 | Pop3 | Pop4 | Pop5 | Pop6 | Pop7 | Pop8 | Pop9 |
| Na | 9.250 | 8.750 | 6.583 | 3.417 | 3.250 | 1.917 | 4.250 | 1.000 | 3.750 |
| Na Freq.>=5% | 6.333 | 5.167 | 4.083 | 3.417 | 3.250 | 1.917 | 4.250 | 1.000 | 3.750 |
| Ne | 4.989 | 4.856 | 4.166 | 3.138 | 2.853 | 1.917 | 3.603 | 1.000 | 3.164 |
| I | 1.734 | 1.685 | 1.467 | 1.051 | 0.935 | 0.635 | 1.314 | 0.000 | 1.149 |
| No. Private Alleles | 1.750 | 1.750 | 0.333 | 0.167 | 0.083 | 0.000 | 0.167 | 0.083 | 0.333 |
| No. LComm Alleles (<=25%) | 1.250 | 1.083 | 0.583 | 0.250 | 0.000 | 0.083 | 0.583 | 0.083 | 0.417 |
| No. LComm Alleles (<=50%) | 3.250 | 3.000 | 2.250 | 0.917 | 0.750 | 0.250 | 1.417 | 0.250 | 1.083 |
| He | 0.735 | 0.722 | 0.671 | 0.579 | 0.500 | 0.458 | 0.694 | 0.000 | 0.616 |
| UHe | 0.744 | 0.734 | 0.685 | 0.631 | 0.545 | 0.611 | 0.747 | 0.000 | 0.672 |
Wright’s F-statistics revealed that the 12 microsatellite markers detected significant population structure. The mean genetic differentiation index (FST) among the parasite populations for all the 12 MS loci was 124.91 reaching statistical significance (p=0.001), Table 3 summarizes this.
Table 2b: Showing mean values across 9 populations.
| Locus | Mean N | Fst | P Fst |
|---|---|---|---|
| 2490 | 125 | 0.227 | 0.151 |
| ARA2 | 125 | 0.257 | 0.085 |
| PFG378 | 125 | 0.159 | 0.531 |
| PFpk2 | 125 | 0.230 | 0.087 |
| Poly | 125 | 0.213 | 0.081 |
| TA108 | 125 | 0.180 | 0.342 |
| TA40 | 124 | 0.431 | 0.004 |
| TA42 | 125 | 0.165 | 0.396 |
| TA60 | 125 | 0.193 | 0.264 |
| TA87 | 125 | 0.260 | 0.023 |
| TA1 | 125 | 0.333 | 0.011 |
| TA81 | 125 | 0.230 | 0.058 |
| Tot | 124.917 | 0.236 | 0.001 |
FST values across the 12 markers with their mean and FST values
N: Total number of isolates, Na: Number of different alleles over pops, FST: Wrights F statistics with12 loci accounting for population structure
Table 3: Showing FST values across 12 loci.
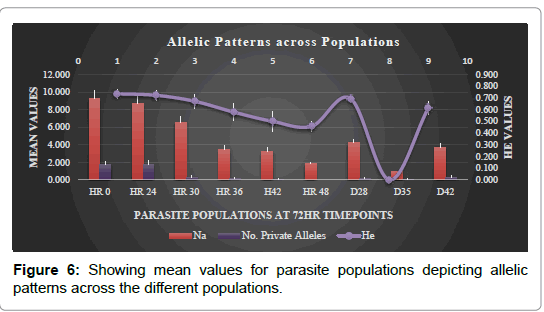
Figure 6: Showing mean values for parasite populations depicting allelic patterns across the different populations.
Multiple copies of genes are associated with over expression of the encoded traits. It has been shown that differential copy numbers and/ or differential transcription of putative drug resistance gene candidate’s influences responses to drugs in malaria parasites. In P. falciparum, multiple Pfmdr1 genes copies have been implicated in resistance to quinine derivatives. These polymorphisms have been previously documented in SEA and shown to confer resistance to mefloquine.
In South East Asia, increased Pfmdr1 copy number has been attributed to prolonged use of Mefloquine. Over the past decade, Kenyan samples show a trend of transition to multiple Pfmdr1 copy number absence of MQ use. Moreover, lumefantrine that is the presently used partner drug for treatment of malaria in Kenya is a different class of antimalarial therefore construing the potential cross resistance upon introduction of the latter for treatment. These results further suggest that it is necessary to understand mechanisms drugs prior to policy recommendation in order to adequately predict co-resistance that would emerge against other non-policy recommended drugs.
Analysis of 118 Kenyan samples collected between 2013 and 2015 showed that 2% had 2 copies of Pfmdr1 gene while an additional 8% had greater than 1.75 copies. Previous analyses of Pfmdr1 gene copy number in Kenya for samples obtained between 2008 and 2011 from the same site showed that all samples had 1 copy of Pfmdr1 gene while a follow-up study including the 2008 to 2013 samples reported Of the 129 sample isolates analyzed, eight carried multiple Pfmdr1 gene copy numbers. Seven of these were post-ACT and one was pre-ACT. The mean Pfmdr1 gene copy number for the eight samples was 1.85 (95% CI, 1.76-1.93) compared to the single Pfmdr1 gene copy number which was 1.21 (95% CI, 1.17-1.24). Previous studies done in Mbita Kenya have proposed that codons 86, 184and 1246 are associated with parasite recurrence after AL [2] with the key determinant being copy number variation. Observation of elevated copy number in this study assessed in 2015 samples show the activity in the Pfmdr1 gene. The increasing Pfmdr1 copy number suggests that lumefantrine resistance could soon emerge and putting the country at risk of Coartem failure.
To determine whether there was change in Pfmdr1 gene copy in Kenyan parasites with the ACT dosing, this study analyzed 118 samples, six samples carried multiple Pfmdr1 gene copy numbers. The mean Pfmdr1 gene copy numbers for the six samples was 1.80 (95% CI, 1.30- 2.20). Significant selection of wild type alleles and multidrug resistance 1 (Pfmdr1) gene at codon 86 followed by P. falciparum chloroquine resistance transporter (Pfcrt) gene at codon 76 occur in recrudescent and re-infecting parasites after AL treatment.
However, since only a small number of samples were tested, additional studies are needed to determine if indeed there is any causal association between these SNPs and artemisinin sensitivity.
Parasite clearance slope half lives
A 6 hourly monitoring of parasitemia showed a gradual decrease across the 72 h period of dosing. Ultimately all parasitemia resolved by day 3 implying that ACT treatment was effective. Non-parametric unpaired t-test (with Welch’s correction) and all the parasites in this study were categorized as fast-clearing (compared to SEA. Parasites were stratified based on the median clearance half-life of 4.00 h. Those above the median half-life were classified as fast clearing and those below the median half-life as faster clearing. Parasite clearance half-life >5 h are regarded as slow clearing infections and are often associated with mutations in Kelch propeller gene that confer resistance.
In SEA, there is a strong relationship between mutations and delayed parasite clearance following artemisinin treatment [16]. However, with the exception of one case in Nigeria and two cases in DRC, there have been no reported cases of delayed parasite clearance in SSA and studies have reported median half-lives not more than 4 h. Notably, one patient in this study had a parasite clearance half-life 5.1 h that did not show association with any polymorphisms analyzed here therefore suggesting that the observed treatment outcomes could not be substantiated by analyses of, Pfubp1, Pfap2mu, Pfcrt and Pfmdr1 polymorphisms or copy number investigated by this study. Similar to the previous findings, this study revealed that the median half-life was 2.55 h, with a range of 4.00-11.05.
Western Kenya is a high transmission area with no reports of delayed parasite clearance to artemisinin treatment. However, the possibility that P. falciparum populations in Kenya may be less responsive to ACT therapy provides a strong rationale for further investigation. Whether the submicroscopic persisting parasite phenotype described in Kenya is related to the more pronounced microscopically detectable slow clearance described from western Cambodia should be the new focus.
Codons 72-76 of Pfcrt
The wild-type Pfcrt allele at codons 72-76 of the CRT protein was present in 115 of the 118 (97%) evaluable parasite isolates prior to treatment (day 0). Laboratory and clinical studies have associated chloroquine resistance with point mutations in the gene Pfcrt therefore causing Plasmodium falciparum chloroquine resistance which is a major cause of worldwide malaria mortality and morbidity.
A recent pooled analysis from clinical trials showed patients infected with parasites carrying the Pfcrt N86 allele and/or increased the Pfmdr1 copy number (as independent risk factors) were at a significantly at a greater risk of AL treatment failure than those whose parasites carried the 86Y allele or a single copy of the Pfmdr1. The prevalence of Pfcrt K76 and N86 increased from 6.4% in 1995 to 93.2% in 2014 and 0.0% in 2003 to 92.4% in 2014. In this study, we saw an increase of the Pfcrt 76T from 92.4% in 2014 which was the lowest reported prevalence, to a high of 97% (n=118). With the prevalence of Pfcrt K76 allele on the rise, suggestive of reemergence of chloroquine-sensitive P. falciparum malaria in western Kenya, there is need to evaluate the role of chloroquine or compounds in the same class in the treatment of malaria.
Further analysis will substantiate suggestions that children with Pfcrt genotypes present prior to treatment is associated with risk of parasite recurrence at day 28 or day 42 [2] and should be a cause to rethink the issue with the deployment of first-line treatment of uncomplicated malaria.
Plasmodium falciparum ubiquitin protein 1
The new candidates Pfubp1 (encoding ubiquitin carboxyl-terminal hydrolase 1) has been implicated in artemisinin resistance in the rodent parasite P. chabaudi, have been shown to have polymorphic homologues in P. falciparum [2]. These polymorphisms have yet to be fully validated as markers in parasites with reduced sensitivity to artemisinin derivatives in vivo. However, genome-wide association studies of P. falciparum isolates from coastal Kenya associated Pfubp1 variant E1528D with reduced susceptibility to artemisinin in vitro were recently. This study found E1528D as the most polymorphic locus in the Pfubp1 at 18%. Unlike previous studies, this mutation did not show association with clearance rate. Further analysis on the subsequent time points revealed increased frequencies to 19% with diversity across the locus implying that mutations around codons 1520-1540 occurred in all populations. These high frequencies did not correlate with treatment outcomes.
Microsatelite
A subset of samples with positive parasitemia beyond 24 h time point was analyzed. The samples depicted infections containing >1 malaria clones having high genetic diversity and high infection complexity. The samples were analyzed for parasite clone composition and had the 12 MS namely 2490, ARA2, PFG378, PFpk2, Poly a, TA 108, TA 40, TA42, TA60, TA87, TA1 and TA81. They were located across the genome in each of the 99% of the samples. The mean number of alleles in all the loci across the 8 populations ranged from 9.250 to 1.000. The mean unbiased HE was 0.672 the mean number of clones per infection was 6 IQR=10.0-2.0 with a 95% CI. This suggested highly polymorphic parasites and alleles widely distributed across the populations indicating a high genetic diversity and infection complexity.
Presence of these in single infection is indicative of multiplicity of infection that could depict selection pressure on the parasite due to either extensive crossing or super infection. COI brought about by the non-random association of alleles has significant implication for the spread of multi-locus drug resistance haplotypes. As a result of high levels of inbreeding there is an increase in their dispersal.
Genotyping of malaria infected blood samples has complicated existence of infections containing >1 malaria clones. Quantifying the complexity of multiplicity of multiclone infection is of particular interest since it can provide information on the force of infection and allows predictions to be made about the potential for re-assortment and recombination in malaria populations.
Studies done in Kenya have shown Kenyan parasite populations have low genetic differentiation. Similarly, our study revealed parasite populations had low genetic differentiation with the overall mean FST of 0.236 and none of the parasite analyzed having matching haplotypes. Further, the study detected 8 populations that ranged between 0.182- 0.000 based on the Shannon diversity index. Importantly, none of the parasite analyzed had matching haplotypes indicating a high genetic variability. Studies conducted in Zimbabwe and the DRC, regions of high malaria transmission, described the parasite population as having high genetic diversity and significant LD.
We would like to thank all the patients, clinical and other support staff at the study sites. We would like to thank all the colleagues in the Malaria Drug Resistance Laboratory for their technical and moral support. We would also thank the Director of KEMRI for permission to publish this work. This work was supported by the Division of Global Emerging Infections Surveillance and Response System Operations (GEIS). The opinions and assertions contained herein are private opinions of the authors and are not to be construed as reflecting the views of the US Army Medical Research Unit-Kenya, the Department of the Army, the Department of Defense or the U.S. Government.
A.N.A executed the project; run the assays, data analysis and manuscript writing. L.J.C, data analysis, project design, running experiments, data analysis B.O, D.J and K.M., run experiments/assays L.J.C, supervised project activities H.M.A, supervised and assisted in data analysis. B. A., was the Principle Investigator of the efficacy study, supervised project and advised on data analysis. H.A, supervised project activities and provided guidance on the analysis plan, conceived and designed the project; data analysis and manuscript writing.