Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Research Article - (2014) Volume 3, Issue 1
Keywords: Lipase, Ammonium sulfate, Purification, Characterization, Kinetic study
The use of enzymes created opportunities for developing a green, sustainable and modern industrial chemistry due to excellent specificity, being atom economic, mild reaction conditions, energysaving process and simplicity. In particular the use of hydrolytic enzymes such as protease, lipase and amylase in the industrial enzyme market is growing steadily.
Lipases (E.C. 3.1.1.3) can be broadly defined as enzymes that catalyze the hydrolysis of ester bonds in substrates such as vitamin esters, phospholipids, triglycerides (TGs) and cholesteryl esters [1]. Interest in lipases has been increasing in the past few years because of the actual and potential uses of this enzyme in applications like hydrolysis of fats and oils, organic synthesis, modification fats, chemical analysis and flavor enhancement in food production. The isolation and purification of lipases is reported in various models, chiefly microorganisms but there is a paucity of information about the isolation and purification from carps. Therefore, the lipases from the tissues of these aquatic vertebrates have received much less attention than those from microorganisms. Hepatopancreas as digestive organs found in the fishes remain unexplored lipolytic enzyme sources that could have unique characteristics. Fish lipases demonstrate novel activities that have potential industrial applications [2]. Purification of lipase from the Indian Major Carps may also be of benefit for the development in the field of aquaculture. Thus the present study intricacies on the isolation purification and characterization of lipase from the digestive gut of Indian Major carp, Catla catla (catla).
Culture of fish
Indian major carp Catla catla (catla) were collected from local fish market, INA, Delhi and were acclimatized at outdoor conditions for 10 days. Fish were fed with artificial diet containing 40% protein. Artificial diet was prepared by using dried fish powder, wheat flour, cod liver oil and vitamin and mineral premixes [3]. Fishes were kept in fasting condition for 48 h before sampling.
Sampling of tissue
Two fishes were anesthetized with MS 222 (Tricaine methanesulfonate) before sacrifice. Individual fish was dissected; digestive system and associated glands (hepatopancreas) were removed from the body, cleaned, weighted and immediately frozen at -20ºC till further use.
Preparation of crude extract
Digestive systems and hepatopancreas collected from two fishes were pooled; total weight of tissue was recorded and homogenized in sample buffer (10 mM Tris-HCl and pH 7.2) in the ratio of 1:3. Homogenized solution was passed through pretreated cheese cloth for the separation of excess fats. The cheese cloth was treated by keeping it in 1% EDTA solution for 12 h. The suspension was then centrifuged for 30 min at 13000 rpm at 4°C. The floating fat phase was removed and the solution was filtered through Buchner funnel (Borosilicate grade-2) under vacuum (ROCKER-300, TARSONS, Mumbai, India), volume of the solution was noted and thus formed solution is called crude extract.
Estimation of protein
Protein concentration was measured by the method of Bradford [4]. Bovine serum albumin was used as a standard.
Assay of lipase activity
Lipase was assayed according to the method of Winkler and Stuckman [5]. The basis of this assay is the colorimetric estimation of para-Nitrophenol (pNP) released as a result of enzymatic hydrolysis of para-Nitrophenyl palmitate (pNPP). The substrate was prepared by dissolving pNPP (30 mg) in 10 ml of isopropanol mixed with 90 ml of 0.05 M Sorenson’s phosphate buffer (pH 8.0) containing 207 mg of sodium deoxycholate and 100 mg of gum arabic. Aliquot (1.2 ml) of the freshly prepared substrate was prewarmed at 37°C and incubated with the enzyme extract of 50 μl for 15 min, at 37°C. Then the absorbance was recorded at 410 nm against enzyme free control. One enzyme unit is defined as 1 nmol of p-Nitrophenol enzymatically released from the substrate ml-1 min-1. The extinction coefficient of para-Nitrophenol is 15, 000 cm2 mg-1.
Purification of crude sample
Crude extract is subjected to ammonium sulfate fractionation [6]. Saturated ammonium sulfate (20%) was slowly added to the crude extracts with constant stirring for 2 h after final addition of ammonium sulfate. Then centrifuged at 13,000 rpm at 4ºC for 30 min and the supernatant was brought to 80% saturation by further addition of ammonium sulfate. Then the sample was centrifuged at 13000 rpm at 4ºC for 30 min and the precipitate was collected; re suspended in the minimal volume of sample buffer. The sample was dialyzed (Sigma, D 0530, 12.4 k Da, St. Louis, USA) overnight against the equilibration buffer (10 mM Tris-HCl and pH 7.2) and was filtered through 0.45 μm Polyethersulfone membrane (25 mm diameter, Whatman, Maidstone, England) syringe filter. Lipase activity and protein concentration of filtrate were assayed. Filtrate was applied slowly (0.5 ml/min) to a DEAE-Cellulose column (0.5×15.0 cm, Bio-Rad, Hercules, CA 94547, USA), which was previously equilibrated with same sample buffer. The column was washed with the equilibration buffer or sample buffer until the effluent had no detectable absorption at 280 nm. The whole process was carried out at 4ºC.
Lipase like enzymes was then eluted from the column using a step gradient of NaCl with different concentrations ranging from 100 mM to 800 mM NaCl in the starting buffer. Finally, the column was washed with 1 M NaCl in the starting buffer. The flow rate was adjusted to 24 ml/h (peristaltic pump, Amersham Biosciences, Uppsala, Sweden) and 5 ml fractions were collected. The eluents were monitored at 280 nm for protein and at 410 nm for lipase activity. Highest activity fractions were pooled as Purified Fraction (PF) and were filtered through Whatman polyethersulfone membrane (0.45 μm). Then the sample was divided into aliquots. Aliquots were used for physical and kinetic characterization. Few lyophilized aliquots were used for molecular characterization.
Characterization of purified sample
Optimum pH: The effect of pH on the enzyme activity was evaluated by measuring the activity at various pH’s, using pNPP at 37°C as the substrate. Range of pH was varied from 3.0 to 10.2 using citrate-phosphate-Tris-glycine buffer of 0.05 M [7]. The substrate at appropriate pH was used as blank, distilled water being added instead of enzyme.
Optimum temperature and thermostability: Optimum temperature of pure lipase activity was measured using pNPP at 37°C as the substrate and varying temperature at the range of 10-80°C.
Pure lipase temperature stability was evaluated by incubation at various temperatures ranging from 20 to 40°C for 60 min and measuring residual activity after 30 min using pNPP as the substrate at 37°C [7].
Effect of CaCl2 on thermal stability: The effect of CaCl2 on thermal stability was determined by heating the enzyme dissolved in 10 mM Tris-HCl and pH 7.2 [8] in the presence of 2, 10 and 20 mM CaCl2, at 40°C for various time intervals (0, 1, 2, 3 and 4 h). At the time designated the samples were assayed for remaining activity with pNPP at 37°C as the substrate (pH 8.0).
Kinetic studies: The activity was assayed with different final concentrations of pNPP at 37°C as the substrate ranging from 0.625 to 20 mM [9]. The final enzyme concentration for the assay was estimated as micro moles (μM).Kinetic parameters including Vmax and Km were evaluated by plotting the data on a Line weaver-Burk double reciprocal graph [9]. Turnover number (Kcat) was calculated from the equation: Kcat=Vmax/[E], where [E] is the active enzyme concentration (μM) and Vmax is the maximal velocity. Catalytic or the physiological efficiency of the substrate was calculated by the equation: Kcat/Km.
SDS-PAGE: Separation of proteins in the enzyme extracts was done by 12% SDS-PAGE according to Laemmli [10]. Enzyme extract (20 μg protein) was loaded onto each well and electrophoresis was performed (30 mA) on a vertical dual mini gel electrophoresis device (Gene-I, Bangalore, India) at a controlled temperature of 4°C. The lipase composition was studied after separation of proteins by substrate SDS-PAGE [11], samples contained 5 m Units of activity was loaded onto each well. The gel was then washed, stained with 0.1% Coomassie brilliant blue (CBB, Mumbai, India) in methanol:acetic acid:water (40:10:40) for 2 h. Destaining was done with the same solution without CBB for 1 h. Protein bands were observed and was observed for the molecular weight determination. The gels were documented in calibrated densitometer (GS-800, Bio-Rad, CA 94547, USA) with help of Quantity one – 4.5.1 software.
Circular dichroism: Circular Dichorism is based on differential absorption of left and right circularly polarised radiation by chromophores which either possess intrinsic chirality or are placed in chiral environment. CD spectrum can be analysed to give the content of regular secondary structure features such as α-helix and β–sheets in far UV region (240-180 nm). In near UV region (320-260 nm), tertiary structure of a protein can be studied [12].
The protein sample was prepared by diluting the protein with sample buffer in 1:1 ratio. The solution was pipetted into the cuvette and the baseline was scanned from 200-260 nm using CD machine (Applied PhotoPhysics, Chirascan™ CD Spectrometer).
Purification of enzyme
Lipase was extracted and concentrated by using ammonium sulfate precipitation in the cut off range 20-80%. Initial specific activity was found to be 406.1 units in the crude extract (Table 1). Resultant precipitate was subjected to dialysis to remove excess salts. Dialysate specific activity was found to be 408.3 with recovery of 42.2%. Dialysate was subjected to purification by using ion exchange chromatography on DEAE-Cellulose. Bound fraction was eluted by using step gradient of sodium chloride in the range of 100-800 mM. An active peak in fraction numbers 48-55 and minor peaks were found in the elution profile of DEAE-Cellulose column (Figure 1).
| Steps | Volume | Protein | Total Protein | Specific Activity | Total Activity | Recovery (Yield) | Fold Purified |
|---|---|---|---|---|---|---|---|
| (ml) | (mg/ml) | (mg) | (Units/mg) | (Units) | (%) | ||
| Crude Extract | 115 | 1.70263 | 195.80263 | 406.1566425 | 79526.5 | 100.0 | 1.0 |
| 20 to 80 % (NH4)2SO4 precipitation | |||||||
| Dialysis | 40 | 2.05658 | 82.2632 | 408.31 | 33588.8 | 42.236 | 1.0053 |
| Ion Exchange Chromatography | |||||||
| PF 1 | 36 | 0.12303 | 4.42 | 1438.72 | 6372 | 8.01242 | 3.542 |
Table 1: Summary of Catla catla lipase purification. The basis of this assay is the colorimetric estimation of para-Nitrophenol (pNP) released as a result of enzymatic hydrolysis of para-Nitrophenyl palmitate (pNPP). One enzyme unit is defined as 1 nmol of p-nitrophenol enzymatically released from the substrate ml-1 min-1. The extinction coefficient of para-nitrophenol is 15000 cm2 mg-1.
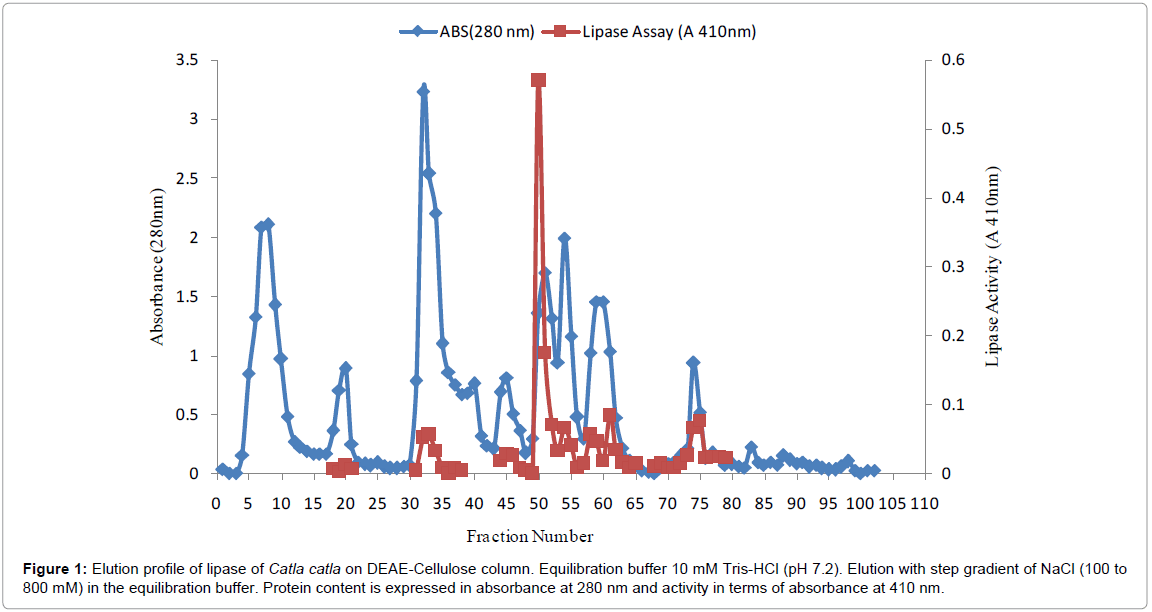
Figure 1: Elution profile of lipase of Catla catla on DEAE-Cellulose column. Equilibration buffer 10 mM Tris-HCl (pH 7.2). Elution with step gradient of NaCl (100 to 800 mM) in the equilibration buffer. Protein content is expressed in absorbance at 280 nm and activity in terms of absorbance at 410 nm.
Active fractions were pooled as Purified Fraction (PF), the final specific activity of purified lipase was found to be 1438.72 units with recovery of 8.01% and purification fold was 3.54.The purified fraction was further processed for physical, kinetic and molecular characterization. Nayak et al. [13] used SEPHADEX G100 Chromatography to purify intestinal lipase from Labeorohita and obtained 3.6 fold purify with a specific activity of 38.4 U/mg. Gorgun and Akpinar [14] purified lipase from the liver of carp Cyprinus carpio by combined PEG-6000 on Q Sepharose on Sepharacryl S200-HR on Phenyl Sepharose CL-4B. 75.5 fold was purified and 90.38 U/mg specific activity was obtained. Purification fold of 34.5 was obtained with 6.21 U/mg specific activity when lipase was purified from the dorsal part of Cirrhinusreba using gel filteration on Sephadex G-50 (F-1) and DEAE Cellulose [15].
Physical characterization
Optimum pH: Effect of pH on lipase activity of the digestive tissue of Catla catla was measured in series of buffer range for pH 2.0 to 10.2 using pNPP as substrate at 37°C. Optimum pH in the study was found to be 7.8 (Figure 2). In the acidic pH at 2.5, the lipase activity was 10% and then it gradually increased up to 75% at pH 4.0. Thereafter activity was gradually decreased; activity was declined by 25% and was 50% at pH 7.0. In the pro alkaline condition, activity increased to maximum (100%) at pH 7.8 and thereafter it decreased finally to 30% at pH 10.0.Triglyceride lipase pH of Gadus morhua was 8.25 [16] using Olive oil as substrate and pH of purified lipase of Cyprinus carpio was 8.0 using the substrate pNPB [14]. These results were in a strong correlation with those of present study. However under acidic conditions, the charge distribution and conformation were changed and enzymes could not bind to substrate properly [17].
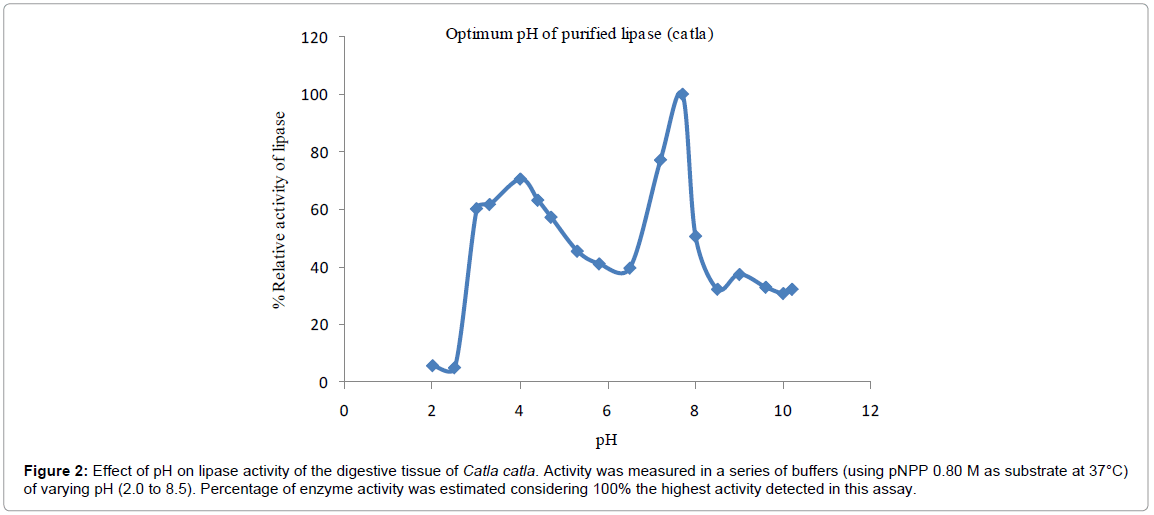
Figure 2: Effect of pH on lipase activity of the digestive tissue of Catla catla. Activity was measured in a series of buffers (using pNPP 0.80 M as substrate at 37°C) of varying pH (2.0 to 8.5). Percentage of enzyme activity was estimated considering 100% the highest activity detected in this assay.
Optimum temperature: Effect of temperature on the hydrolysis of pNPP of lipase is shown in Figure 3. Optimum temperature was found to be at 20°C. Activity was found to be 60% at 10°C and then it increase by 40% with maximum activity (100%) at 20°C. Therefore for every rise in the temperature, lipase activity gradually decreased with final activity of 10% at 80°C. In the present study lipase showed optimum activity at 20°C.
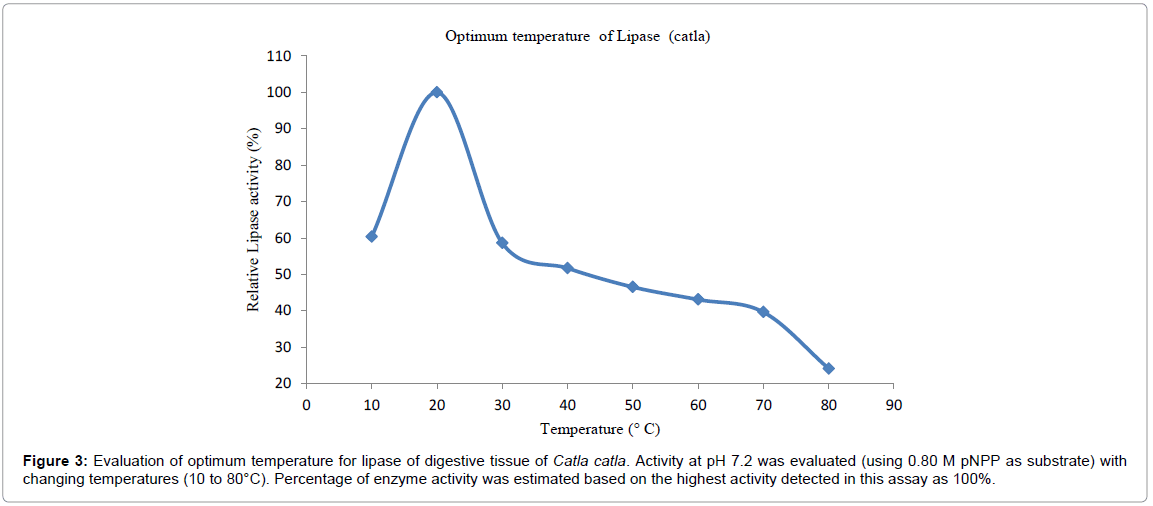
Figure 3: Evaluation of optimum temperature for lipase of digestive tissue of Catla catla. Activity at pH 7.2 was evaluated (using 0.80 M pNPP as substrate) with changing temperatures (10 to 80°C). Percentage of enzyme activity was estimated based on the highest activity detected in this assay as 100%.
Optimum temperature was found to be 15°C for O. mykiss liver lipase [18], 37°C in Cyprinuscarpio [14], 25°C to 30° C for Gadusmorhua digestive lipase [16] and 35°C for C. reba dorsal part lipase [15].
Melting temperature: Melting temperature of purified lipase of Catla catla was evaluated by using the relative activity of lipase at various temperatures by using pNPP as the substrate. Melting temperature (Tm) of catla lipase was 42°C (Figure 4) as exactly 50% of the activity lost at this particular temperature.
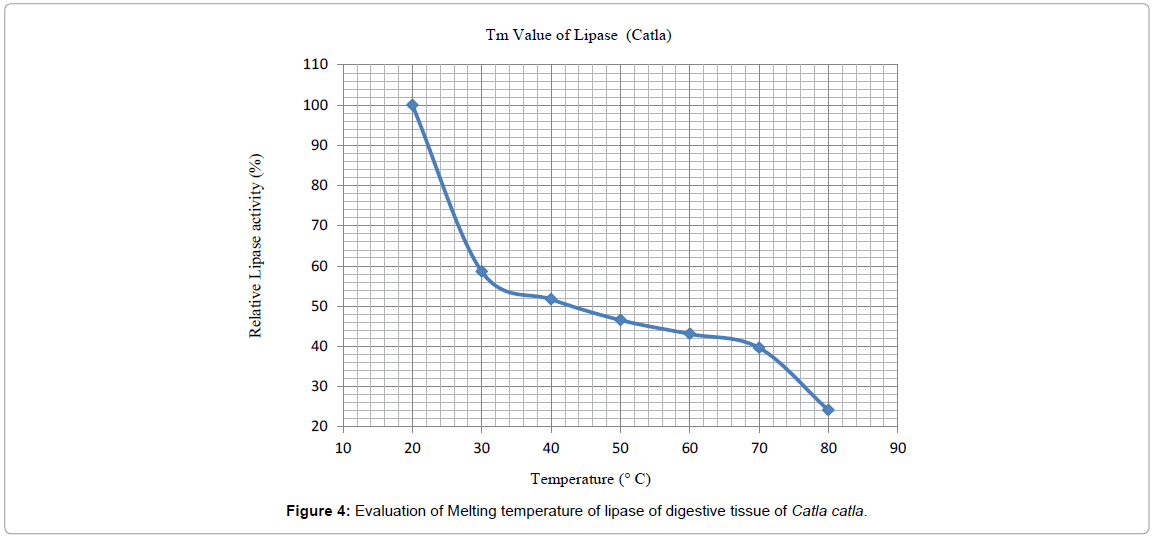
Figure 4: Evaluation of Melting temperature of lipase of digestive tissue of Catla catla.
Activation energy: Arrhenius Activation energy of purified lipase from Catla catla was determined by using the Ln relative activity. The activation energy was calculated as 34.82 KJ/mol/K (Figure 5). The activation energy of cyanobacterial lipase from Spirulina platensis was calculated by using Arrhenius equation and it was found as 146.34 kJ/ mol [19].
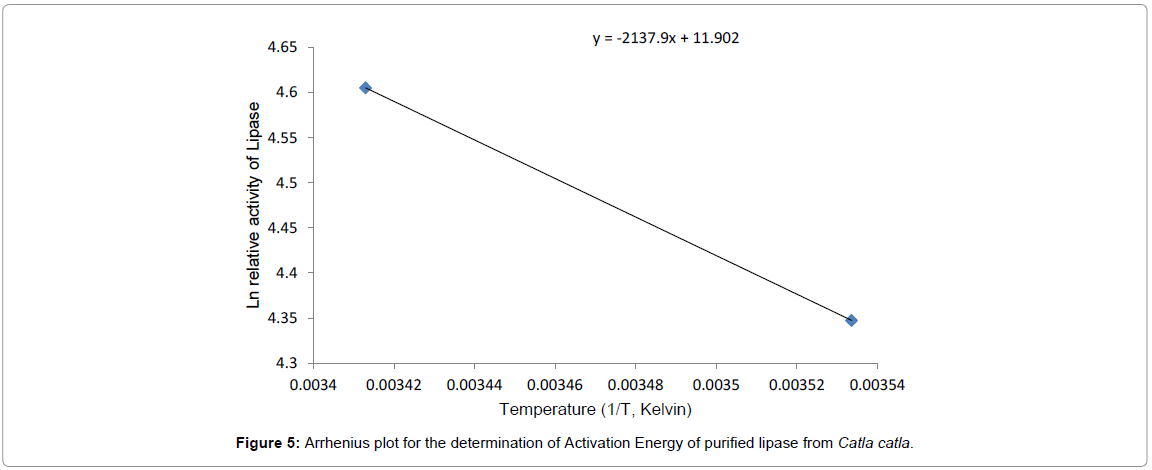
Figure 5: Arrhenius plot for the determination of Activation Energy of purified lipase from Catla catla.
Temperature stability: A 60.2% lipase activity was recorded at 30°C with incubation period of 30 min (Figure 6) and was stable after 60 min of incubation retaining 55% activity. Similar trend was observed at 40°C with incubation period of 30 min and thereafter it decreased to 10% after 60 minutes of incubation showing less stability. Half of the activity was found at 30°C after 30 minutes of incubation and then it slightly decreased to 25% final activity after 60 min of incubation.
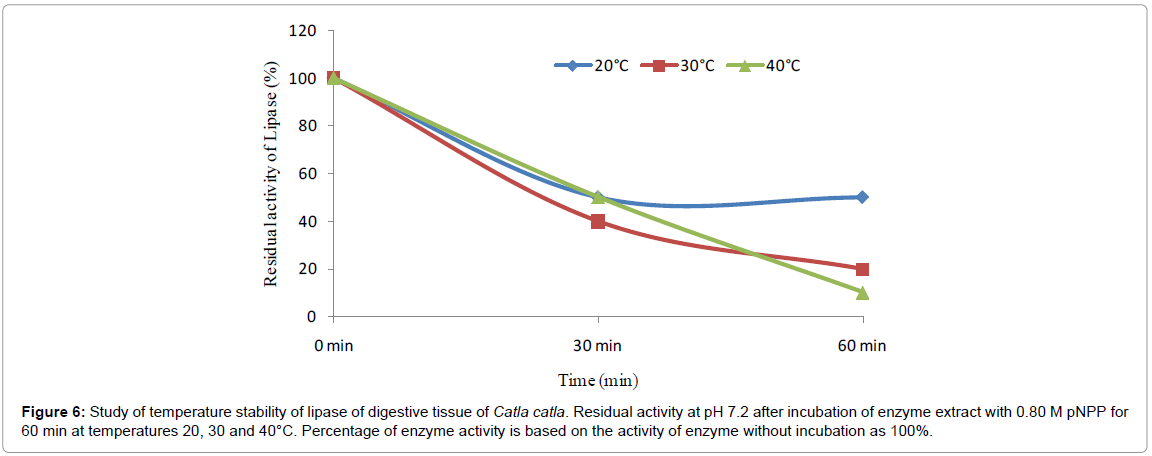
Figure 6: Study of temperature stability of lipase of digestive tissue of Catla catla. Residual activity at pH 7.2 after incubation of enzyme extract with 0.80 M pNPP for 60 min at temperatures 20, 30 and 40°C. Percentage of enzyme activity is based on the activity of enzyme without incubation as 100%.
Residual activity of lipase of Catla catla was stable at temperature 20°C up to 60 minutes of incubation whereas residual activity of lipase gradually declined at 30°C and 40°C. The enzyme activity was clarified to be stable within the temperature range of 30-60°C for C. reba dorsal part lipase [15].
Kinetic study
Kinetic constants Km and Kcat for lipase from digestive gut or hepatopancreas of Catla catla were calculated using Michaelis-Menten and Line weaver-Burk plot (Figures 7a and 7b) and were shown in Table 2. Km and Kcat for the pure lipase of Catla catla were found to be 6.695 mM and 0.0022 s-1, respectively. Catalytic efficiency of lipase was 0.0003412 s-1 mM-1.
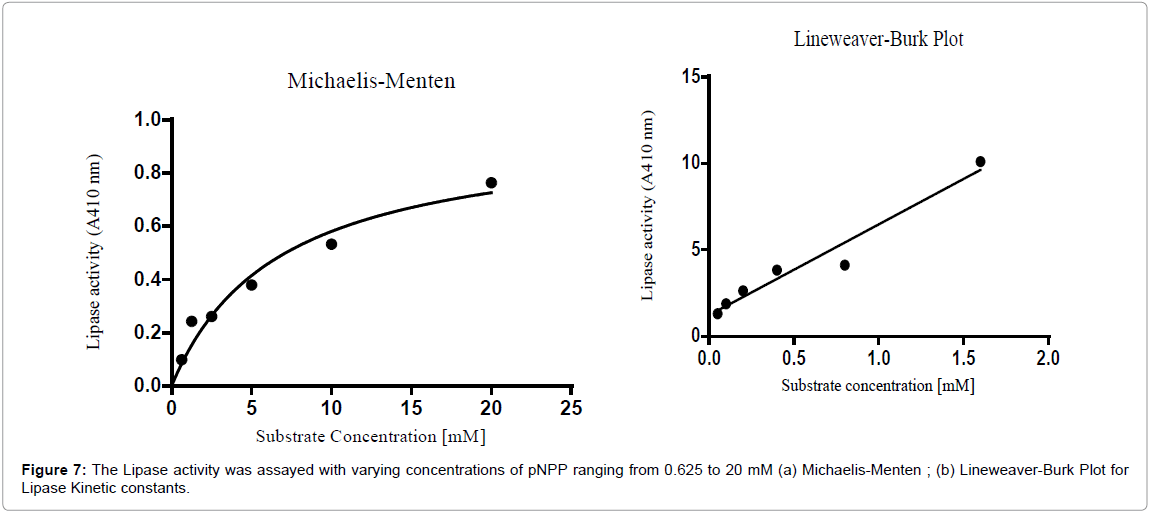
Figure 7: The Lipase activity was assayed with varying concentrations of pNPP ranging from 0.625 to 20 mM (a) Michaelis-Menten ; (b) Lineweaver-Burk Plot for Lipase Kinetic constants.
| Lipase | Michaelis constant, Km (mM) | Turnover number, Kcat (s-1) | Catalytic efficiency, Kcat/Km (s-1 mM-1) |
|---|---|---|---|
| Catla catla (catla)* | 6.695 | 0.0022 | 0.0003412 |
Table 2: Study of kinetic constants of Lipase (Catla catla).
Using different substrates, it appears that lipases purified from different tissues of fish have exhibited different Km and Vmax values. Km and Vmax values for Cyprinus carpio [14] liver lipase were 0.17 mM and 2.6 μmol/ml.dk using p-NPB as substrate respectively. Harmon et al. [20] found the Km and Vmax values as 0.28 mM and 0.016 nmol/h/mg, using tributyrin for O. mykiss liver lipase. Another study focused on liver lipase of O. mykiss determined Km and Vmax values as 0.12 mM and 0.40 U/mg, respectively, using p-Nitrophenyl acetate (p-NPA) as substrate [21]. Aryee et al. [22] indicated that medium and long chain p-Nitrophenyl substrates were useful substrates for the lipase purified from the viscera of M. cephalus. Using p-Nitrophenyl palmitate (p-NPP) as a substrate, they calculated Km and Vmax as 0.22 mM and 20 μ min-1 mg-1.
The catalytic efficiency (kcat/Km) of lipase was found to be 1.5×106 M-1 s-1. These results showed that Km value of the lipase (p-NPP as substrate) from S. platensis was appreciably lower than Km values of lipase from other sources such as A. niger F044 (7.37 mM) [23], Bacillus stearothermophilus MC 7 (0.33 mM) [24] and Bacillus sp.J33 (2.5 mM) [25].
Effect of calcium
The Effect of Calcium chloride on the stability of lipase from Catla catla was examined at pH 7.2 and 37°C (Figure 8). Lipase activity increased by 8.2 times in 2 mM Calcium chloride till 2 hours and thereafter it decreased at the end of 4 hours of incubation. Similarly, lipase activity increased by 6.2 times in 10 mM Calcium chloride till 2 hours and thereafter decreased to initial value at the end of 4 hours of incubation. Its maximum activity increased 8.5 times in 20 mM calcium chloride till 1 hour and then it gradually decreased to the initial value at the end of 4 hours of incubation. Incubation of purified trypsin with 2 mM CaCl2 showed an increase in the activity up to 8 h. The enzyme activity was 11.7% higher after 4 and 8 h of incubation compared to the initial [26].
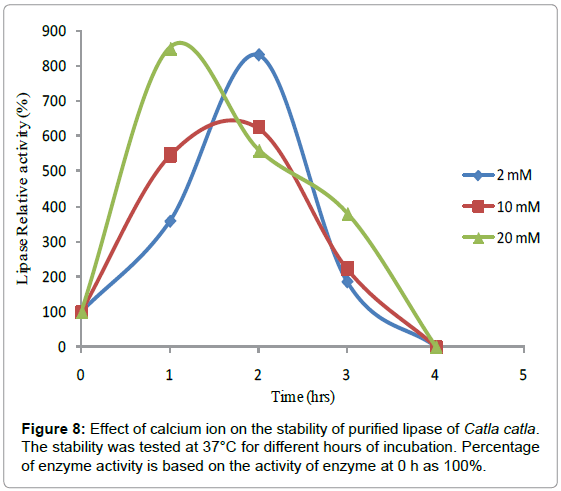
Figure 8: Effect of calcium ion on the stability of purified lipase of Catla catla. The stability was tested at 37°C for different hours of incubation. Percentage of enzyme activity is based on the activity of enzyme at 0 h as 100%.
SDS-PAGE
The SDS-PAGE of purified lipase is shown in Figure 9. A commercial product of low molecular weight marker was (Bio rad) was loaded for comparison. In the crude extract there were three bands with molecular weight 89, 68 and 32 kDa. Two protein bands with molecular weight 30 and 67 kDa were observed in dialysate fraction. Protein bands were purified and reduced to a single homogenate band with 70 kDa molecular weight in ion exchange chromatography, DEAE-Cellulose. Other studies found the molecular weight as 74 kDa for liver lipase of C. carpio [14], 87 kDa for lipase from the dorsal part of Cirrhinus reba [15], 40-43 kDa for O. mykiss liver lipase [20], 43 kDa for S. aurita digestive lipase [27]. As can be seen from these results, we may conclude that lipases have different molecular weight depending on the tissue and the fish species under investigation.
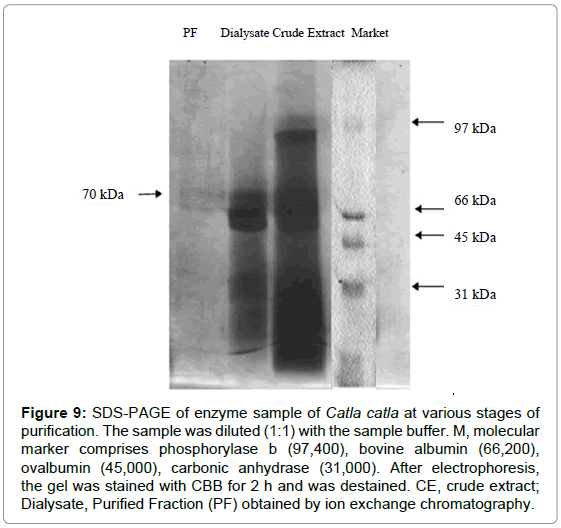
Figure 9: SDS-PAGE of enzyme sample of Catla catla at various stages of purification. The sample was diluted (1:1) with the sample buffer. M, molecular marker comprises phosphorylase b (97,400), bovine albumin (66,200),ovalbumin (45,000), carbonic anhydrase (31,000). After electrophoresis, the gel was stained with CBB for 2 h and was destained. CE, crude extract; Dialysate, Purified Fraction (PF) obtained by ion exchange chromatography.
Circular dichroism
The data was analyzed using K2D2 software. The CD spectra (Figure 10) obtained in the UV range 200-260 nm suggest that the secondary structural arrangement of lipase contains 48.51% of α helices, 9.74% β-sheets and 41.75% random conformations.
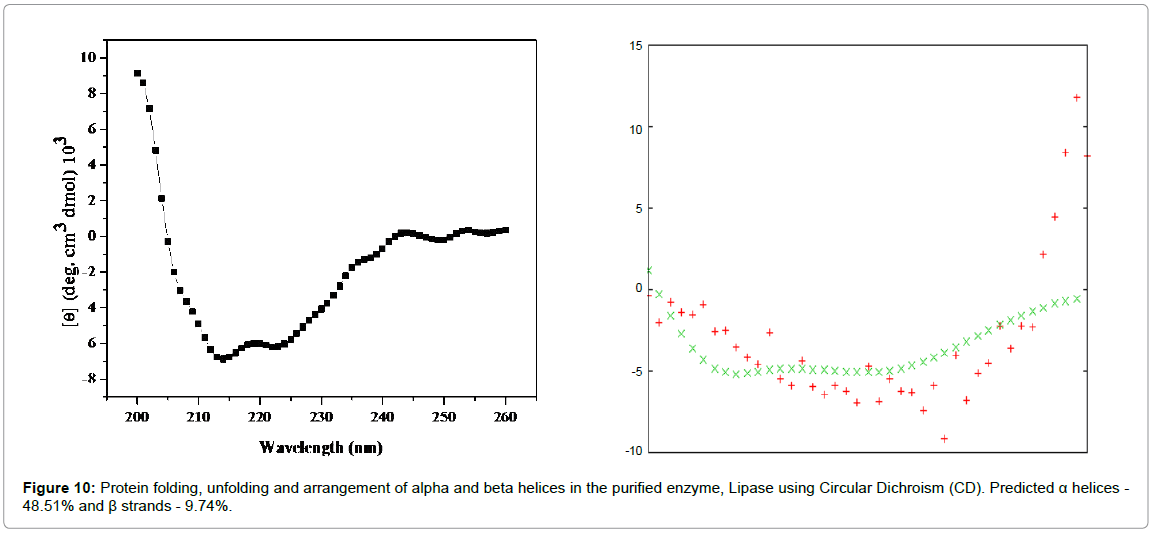
Figure 10: Protein folding, unfolding and arrangement of alpha and beta helices in the purified enzyme, Lipase using Circular Dichroism (CD). Predicted α helices - 48.51% and β strands - 9.74%.
The CD spectra of secondary structure analysis of thermostable lipase from Bacillus sp. showed that the lipase structure contained 38.6% α-helices, 2.2% ß-strands, 23.6% turns and 35.6% random conformations [28].
Authors are thankful to Department of Biotechnology for the financial support and to Dr. P. Hemalatha Reddy, Principal, Sri Venkateswara College, University of Delhi for availing the lab facilities. Circular Dichroism was conducted at Advanced Instrumentation Research Facility (AIRF) JNU, New Delhi.
Experimental set up and data has been collected by Neelima Boora and Prasidhi Tyagi. Paper was compiled and written by Kameshwar Sharma.