Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 2, Issue 1
Although septic encephalopathy leads to be the devastating neurological symptoms including sensory dysfunction, cognitive impairment and unconsciousness, potent substrates and their effects inducing the synaptic dysfunction remain obscure. In this study, we successfully characterized the sensory dysfunction with immunohistochemistry, immunoblotting and electrophysiology. A mouse model of septic encephalopathy was examined at 20 hrs after cecal ligation and puncture or intraperitoneal injection of lipopolysaccharide (1 mg). We found several effects of active enzyme of matrix metalloproteinases-9 (active MMP-9) on the somatosensory cortex, thalamus and prefrontal cortex related to the sensory functions in septic encephalopathy. At first, active MMP-9 was up-regulated. Second, both of the occludin, tight junction protein composing blood brain barrier, and the connexin-43, transmembrane protein of gap junction, which were potent substrate of active MMP-9, were disrupted. Third, the evoked local field potentials in cortical and thalamic neurons were impeded during sensory neuronal stimulation. Conversely, matrix metalloproteinase inhibitor GM6001 significantly protected the reduction of occludin, connexin-43 and the regression of neuronal activities. In conclusion, MMP-9 is a prerequisite candidate for protection of the junction proteins reduction and for the potent therapeutics in the sensory dysfunction in septic encephalopathy.
<Keywords: Prefrontal cortex; Somatosensory cortex; Thalamus; Local field potential; Cecal ligation and puncture; Connexin
SIRS: Systemic Inflammatory Response Syndrome; SE: Septic Encephalopathy; PFC: Prefrontal Cortex; SSC: Somato Somatosensory Cortex; MMP: Matrix Metalloproteinase; LPS: Lipopolysaccharide; LFP: Local Field Potential; BBB: Blood Brain Barrier; CLP: Cecal Ligation and Puncture
Sepsis is a devastating medical condition characterized by systematic inflammatory response syndrome (SIRS). It has been suggested that sepsis results in multiple-organ dysfunction syndrome in response to the processes: 1) endotoxemia (i.e., drastic increase of endotoxin in the blood), 2) SIRS and 3) septic encephalopathy (SE) [1]. SE patients show mental confusion, sensory neuronal impairment and long-term cognitive impairment [2,3]. Patho-physiological mechanisms of brain dysfunction caused by sepsis provoke the development of medical intervention against SE.
Sensory neurons are responsible for converting various external stimuli into corresponding internal stimuli. The stimuli are projected from peripheral neurons and transmitted to thalamus, somatosensory cortex and then integrated at Prefrontal Cortex (PFC) in the central nervous system. These processes participate in the sensory informational processing such as pain generation [4,5]. When the sensory neurons are impaired, the patho-physiology is destined to be peripheral nerve injury such as poly-neuropathy found in the severe septic patients [6]. On the other hand, several reports suggest the PFC-relevant functional impairment in the encephalopathy [7,8]. SE showed the cognitive impairment (i.e. conscious disturbance) in human [9]. In addition, memory deficit with neuronal loss (mainly related to the Central Nervous System (CNS)) [10] and the phrenic nerve neurophathy (Peripheral Nervous System (PNS)) [11] are observed in rodent studies. Hence, the cause of neuronal dysfunctions seems to be attributed to the impairment of informational processing between CNS and PNS.
Connexin, gap junction protein, plays a pivotal role on sensory neuronal function such as vision [12], spinal synaptic transmission [13] and pain [14]. Conversely, loss of connexin results in the sensory dysfunction [14,15]. In the inflammatory symptoms, connexin is often targeted by inflammatory mediator such as interleukin-1β (IL-1β) and tumor necrosis factor-α and lead to be loss of functions [16]. In addition, we previously found that the IL-1β aggravates the synaptic function in the septic brain [17]. In the present study, we hypothesize that sepsis leads to the impairment of substrate such as connexin and the dysfunction of neuronal information processing relevant to sensory function in the septic brain.
Conversely, several evidences suggest that the increased Matrix Metalloproteinases (MMPs) during systemic inflammation play major roles on the functional disturbance in sepsis [18-20]. MMPs which hold super-family are zinc-dependent endopeptidase. MMPs degrade cell-adhesion molecules constructing various proteins localized mainly on cellular surface. The MMPs are initially synthesized as inactive zymogen (i.e., MMPs precursors: pro-MMPs). The pro-MMPs contain pro-peptide domain at carboxy-terminals for keeping their enzymes inactive. Following the removal of pro-peptide domain, pro-MMPs are allowed to be secreted to extracellular milieu as active-MMPs. The broad spectrum inhibitor (e.g. GM6001) antagonizes the active MMPs. On inflammatory symptoms, MMP-9, gelatinase, is especially activated and the active MMP disrupts cell adhesion molecules, tight junction and gap junction proteins. For example, the disruption of tight junction proteins increases the permeability through the blood brain barrier [21]. In a cardiac dysfunction, MMPs impairs the connexin (i.e., gap junction protein) [22]. In the patho-physiology of central nervous system, MMPs aggravate cerebral ischemia and edema [23,24], which are also often found as the complicated symptoms of sepsis [25]. Thereby, it seems that MMP-9 may be a critical enzyme for the synaptic dysfunction in a septic brain. Hence, the regulation of MMP-9 may be critical to prevent the systemic inflammation related to the brain dysfunction in sepsis.
In the present study, we examined whether the MMP-9 exerted the disruption of connexin-43 (i.e., a marker for gap junction [26] and impairment of sensory neuronal function in septic encephalopathy. Our findings suggest the novel molecular mechanisms and therapeutic strategies for the patho-physiology in the septic brain.
Surgery and drug treatment
C57BL6 male mice (8 weeks old) were used for the study. Mice were purchased from Nihon SLC (Hamamatsu, Japan). The surgical procedures were approved by the Animal Care and Use Committee of Osaka University Medical School. Mice were deeply anesthetized by the intra-peritoneal (i.p.) injection of sodium pentobarbital (45 mg/ kg) or urethane cocktail including urethane (0.7 g/kg) and α-chloralose (0.06 g/kg). Body temperature of the mice was maintained at 37°C by a thermostatically-controlled electrical heating pad during surgery. Sepsis was induced by an i.p. injection of lipopolysaccharide (LPS) (5 mg, SIGMA-aldrich, St Louis, MO) (n=7) or cecal ligation and puncture (CLP) (n=6) according to the method described elsewhere [17]. Then, electrophysiological recordings from PFC, somatosensory cortex and thalamus were performed at 20 hrs after the induction of sepsis. In sham-operated mice (n=8), intraperitoneal injection of saline, or similar surgical procedure without CLP were followed. GM6001 was administered by an i.p. injection at 1 hr after LPS- or CLP-treatment. After the conclusion of the experiments, mice were euthanized with excess amount of i.p. injection of pentobarbital (450 mg/kg).
Immunohistochemical staining
Experimental procedures for the immunohistochemical staining of sliced brain samples are described in previous studies [17]. In brief, brain tissues were obtained from the mice at 20 hr after the sham, CLP and LPS-treatments. They were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH=7.2) and provided with the frozen 10 μm thick coronal sections. The primary antibodies used were anti-active MMP-9 (dilution: 1:200, rabbit polyclonal; Abcam, Cambridge, MA) for active enzyme of MMP-9, anti-connexin-43 (dilution: 1:75, rabbit polyclonal; Cell Signaling Technology, Danvers, MA) for gap junction, anti-occludin (dilution: 1:200, rabbit polyclonal; Zymed, San Francisco, CA) for the BBB. Secondary antibodies used included Alexa Fluor 488 goat antimouse IgG (dilution: 1:100, Invitrogen, Carlsbad, CA) and Alexa Fluor 555 goat anti-rabbit IgG (dilution: 1:200, Invitrogen, Carlsbad, CA). The images were recorded with a fluorescent microscope (Axioplan, Zeiss, Oberkochen, Germany). The number of immunoreactive cells in each slice was determined with Image-J software.
Immunoblot assay
Tissues samples were collected from sham or LPS, CLP treated mice and homogenized in the lysis buffer containing 50 mM Tris- HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 10% glycerol, 0.5% SDS, 1.5 mM MgCl2, 1 mM EDTA and the protease inhibitor cocktail (03969-21, Nacalai tesque, Kyoto, JAPAN). The amount of protein in each sample was determined by standard BCA assay (Thermo Fisher Scientific, Waltham, MA). The proteins were separated by SDS-PAGE and analyzed by immunoblot assay as described previously [17]. The primary antibodies used were anti-MMP-9 (dilution: 1:1000), anticonnexin- 43 (dilution: 1:1000) and anti-occludin (dilution: 1:250). Immunoreactive band intensities were analyzed by Image-J software.
Electrophysiology
The anesthetized mice were fixed on a stereotaxic flame (Narishige, Japan). A small hole on cranial skull was opened above the PFC region (coordinates: 3.0 mm anterior from bregma, 1.8 mm lateral, 1.0 mm depth on right hemisphere), somatosensory cortex (coordinates: 2.8 mm lateral from bregma, 1.0 mm depth on right hemisphere), and thalamus (coordinates: 2.3 mm posterior from bregma, 1.3 mm lateral, 3.5 mm depth on right hemisphere) by mechanical drill (minimo power pack, Minitor, Tokyo, Japan). Mice of which brain tissues were injured by the drill were excluded from the analyzed data. Local Field Potentials (LFPs) were recorded by a monopolar tungsten electrode coated with silicon (impedance: 10-20 KΩ), whose tip (length: 10-20 μm) was excised, placed into 800-1000 μm depth from the pial surface of PFC through a hole of cranium. LFPs were amplified (×10000), filtered (lowpass: 100Hz, highpass: 10 kHz) by NEURO DATA ER98 (Cygnus technology, Southport, NC). The data were transferred to Powerlab (AD instruments, Colorado Springs, CO) and recorded into PC for online and offline analysis using scope software (AD instruments, Colorado Springs, CO). Background electrical noises (i.e., artifact) were recorded with an electrode outside the brain during experiments and postmortem excluded from recording data. Sampling rates of the data were set on 1260 Hz.
Spinal nerve transection
The spinal nerve transection was performed in accordance with the method described previously [27]. This experiment was conducted in order to examine whether the evoked-LFPs were activated by sensory neuronal stimulation. In brief, under a deep anesthesia in mice, the spinal nerves on the lumber segment were transected to remove the nerve impulse conduction from a foot pad to the brain. Finally, the wound was closed with nylon sutures.
Statistical analysis
Values are shown as means ± s.e.m. Statistical significance of all differences was assessed by Dunnet-test. For all statistical analyses in the present study, a value of P<0.05 indicates statistical significance.
Immunohistochemical and immunoblot analyses
Up-regulation of active MMP-9 in SE brain: Immunohistochemical staining was conducted to examine active MMP-9 expression in the prefrontal cortex (PFC), somatosensory cortex and thalamic slices (Figure 1A). In sham-operated mice, active MMP-9 immuno-reactivity was barely found in the PFC, somatosensory cortex (Somato) and thalamus (Thalamus) (Figure 1B, left panels). Conversely, in LPS- (Figure 1B, middle) and CLP-treated mice (Figure 1B, right), active MMP-9 immuno-reactivity (red) was robustly found respectively. In GM6001-treated (an inhibitor of metalloproteinase activity) group, the active MMP-9 immuno-reactivity was clearly suppressed even in SE group (Figure 1C). These results suggest that SE induces increase in the active MMP-9 expression in PFC, somatosensory cortex and thalamus.
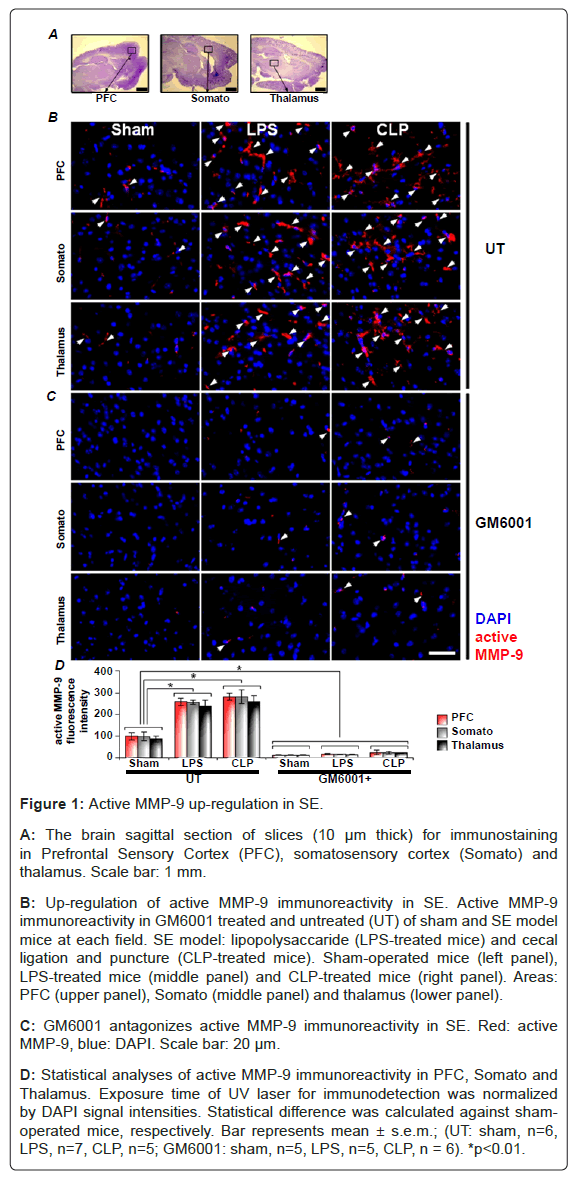
Figure 1: Active MMP-9 up-regulation in SE.
A: The brain sagittal section of slices (10 μm thick) for immunostaining in Prefrontal Sensory Cortex (PFC), somatosensory cortex (Somato) and thalamus. Scale bar: 1 mm.
B: Up-regulation of active MMP-9 immunoreactivity in SE. Active MMP-9 immunoreactivity in GM6001 treated and untreated (UT) of sham and SE model mice at each field. SE model: lipopolysaccaride (LPS-treated mice) and cecal ligation and puncture (CLP-treated mice). Sham-operated mice (left panel), LPS-treated mice (middle panel) and CLP-treated mice (right panel). Areas: PFC (upper panel), Somato (middle panel) and thalamus (lower panel).
C: GM6001 antagonizes active MMP-9 immunoreactivity in SE. Red: active MMP-9, blue: DAPI. Scale bar: 20 μm.
D: Statistical analyses of active MMP-9 immunoreactivity in PFC, Somato and Thalamus. Exposure time of UV laser for immunodetection was normalized by DAPI signal intensities. Statistical difference was calculated against sham-operated mice, respectively. Bar represents mean ± s.e.m.; (UT: sham, n=6, LPS, n=7, CLP, n=5; GM6001: sham, n=5, LPS, n=5, CLP, n = 6). *p<0.01.
MMP-9 impairs BBB in SE
Next, we examine a pathological state for BBB in SE. First, we found the immuno-reactive positive cells for BBB in sham-operated mice (Figure 2). Next, SE was induced with LPS or CLP. Positive immunoreactivity of occludin (i.e., a marker of the BBB) was significantly reduced in PFC, somato and thalamus in the brain slices from LPSand CLP-treated mice (Figure 2A). In addition, the signal intensities of occludin with administration of GM6001 were as the same level as the sham-operated mice (Figure 2B and 2C). These findings suggest that BBB are impaired via up-regulated active MMP-9 in SE.
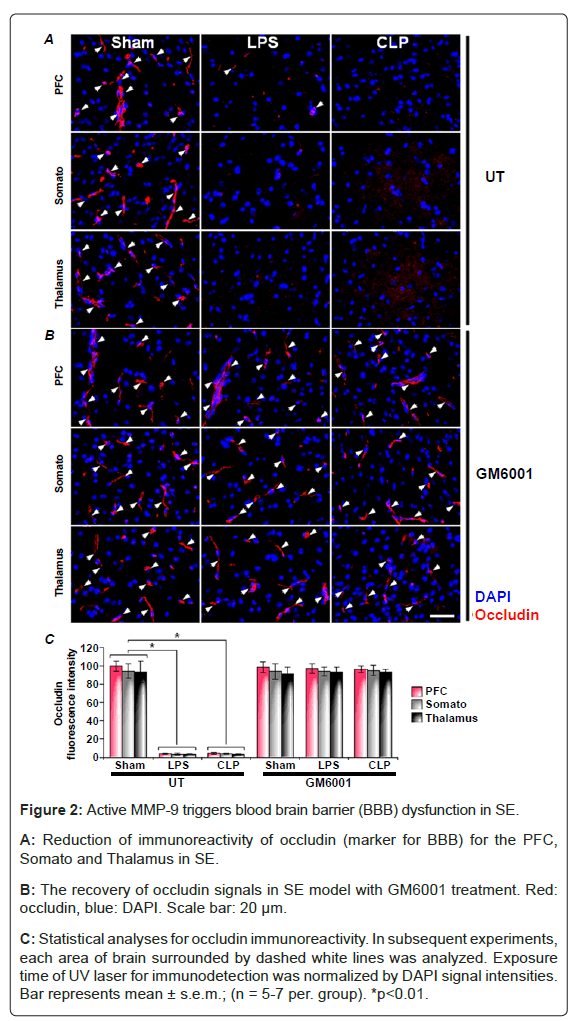
Figure 2: Active MMP-9 triggers blood brain barrier (BBB) dysfunction in SE.
A: Reduction of immunoreactivity of occludin (marker for BBB) for the PFC, Somato and Thalamus in SE.
B: The recovery of occludin signals in SE model with GM6001 treatment. Red: occludin, blue: DAPI. Scale bar: 20 μm.
C: Statistical analyses for occludin immunoreactivity. In subsequent experiments, each area of brain surrounded by dashed white lines was analyzed. Exposure time of UV laser for immunodetection was normalized by DAPI signal intensities. Bar represents mean ± s.e.m.; (n = 5-7 per. group). *p<0.01.
MMP enhances the reduction of gap junction in SE
To determine whether the electrical synapse dysfunction is occurred in SE, we measured immuno-reactivity of connexin-43 (a marker for gap junctions) which is abundantly expressed as hemichannel in the brain [28]. At first, we found that the connexin-43 was predominantly expressed in the Glial Fibrillary Acidic Protein (GFAP) positive cells (Figure 3). Next, in sham-operated mice, the immuno-positive cells of connexin-43 were found in PFC, somato and thalamus of the brain (Figure 4). Whereas, the number of connexin-43 imuuno-positive cells were significantly reduced in CLP and LPS-treated mice (Figure 4A). Conversely, GM6001 canceled the reduction of connexin-43 (Figure 4B). These findings suggest that gap junction is impaired via MMP-9 in SE.
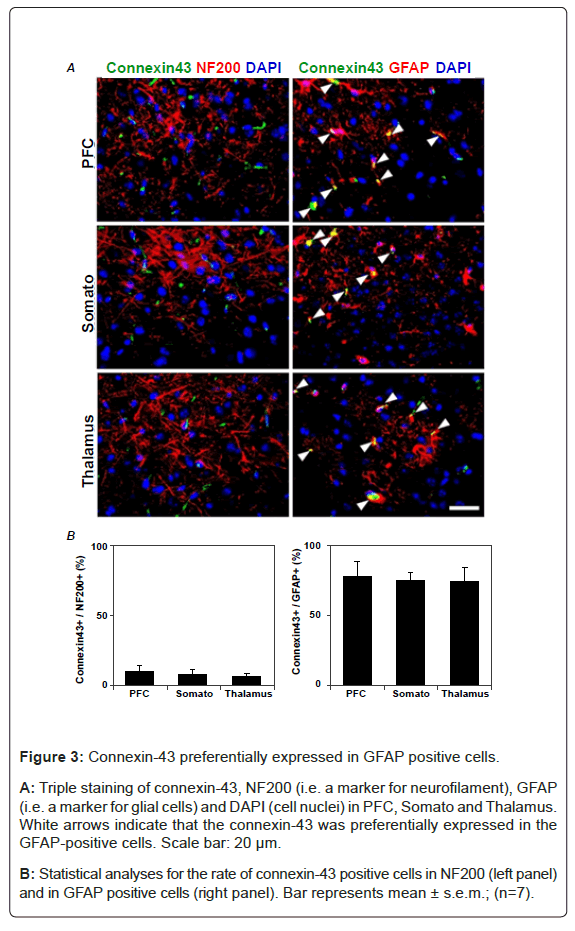
Figure 3: Connexin-43 preferentially expressed in GFAP positive cells.
A: Triple staining of connexin-43, NF200 (i.e. a marker for neurofilament), GFAP (i.e. a marker for glial cells) and DAPI (cell nuclei) in PFC, Somato and Thalamus. White arrows indicate that the connexin-43 was preferentially expressed in the GFAP-positive cells. Scale bar: 20 μm.
B: Statistical analyses for the rate of connexin-43 positive cells in NF200 (left panel) and in GFAP positive cells (right panel). Bar represents mean ± s.e.m.; (n=7).
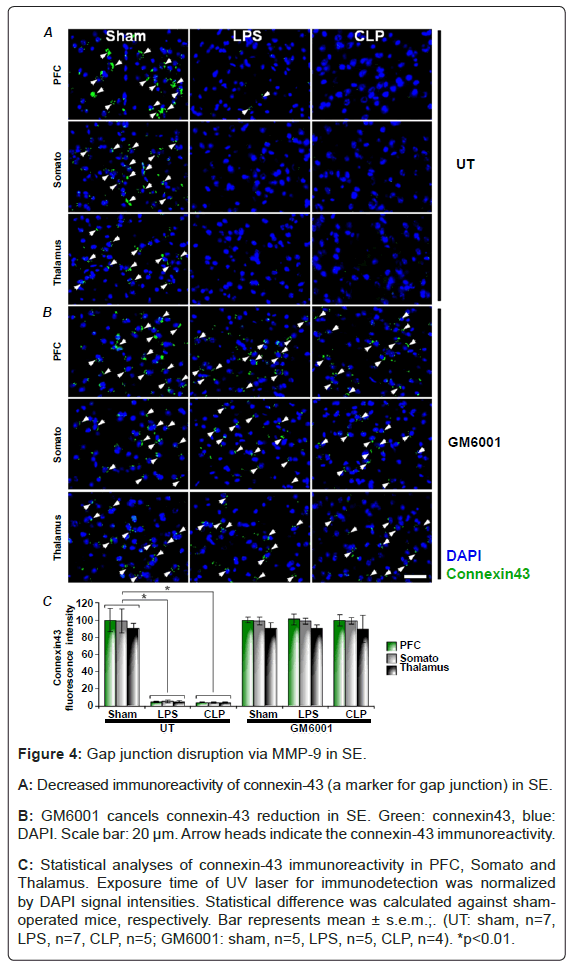
Figure 4: Gap junction disruption via MMP-9 in SE.
A: Decreased immunoreactivity of connexin-43 (a marker for gap junction) in SE.
B: GM6001 cancels connexin-43 reduction in SE. Green: connexin43, blue: DAPI. Scale bar: 20 μm. Arrow heads indicate the connexin-43 immunoreactivity.
C: Statistical analyses of connexin-43 immunoreactivity in PFC, Somato and Thalamus. Exposure time of UV laser for immunodetection was normalized by DAPI signal intensities. Statistical difference was calculated against sham-operated mice, respectively. Bar represents mean ± s.e.m.;. (UT: sham, n=7, LPS, n=7, CLP, n=5; GM6001: sham, n=5, LPS, n=5, CLP, n=4). *p<0.01.
SE results in the protein reduction
To determine whether these immunohistochemical results after LPS and CLP with or without GM6001 are caused by altered protein expression levels or aberrant distribution of immuno-positive cells in the brain, we performed immunoblot assay (i.e., western blotting). As a result, decreased protein expression levels after LPS- and CLPtreatments with GM6001 (Figures 5A-5C) were similar to the results of immuno-histochemical assays (Figures 1, 2 and 4). These results suggest that the protein reductions occur with LPS- and CLP-treatments, and GM6001 effectively inhibits their reductions.
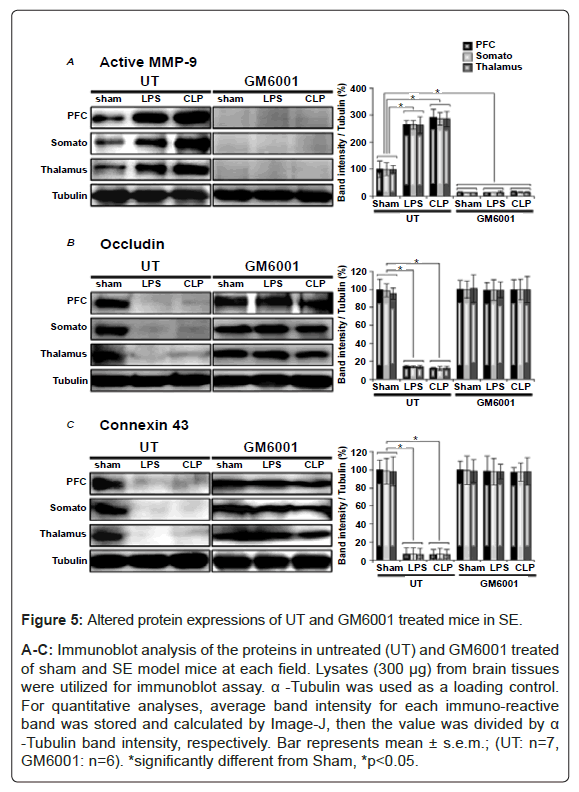
Figure 5: Altered protein expressions of UT and GM6001 treated mice in SE.
A-C: Immunoblot analysis of the proteins in untreated (UT) and GM6001 treated of sham and SE model mice at each field. Lysates (300 μg) from brain tissues were utilized for immunoblot assay. α -Tubulin was used as a loading control. For quantitative analyses, average band intensity for each immuno-reactive band was stored and calculated by Image-J, then the value was divided by α -Tubulin band intensity, respectively. Bar represents mean ± s.e.m.; (UT: n=7, GM6001: n=6). *significantly different from Sham, *p<0.05.
Electrophysiological assays
Evoked local field potentials during a sensory stimulation: To examine the sensory neuronal function, we used a monopolar tungsten electrode to record Local Field Potentials (LFPs) in the PFC, Somato and Thalamus region of mouse brain during paw pressure stimulation (Figures 6A and 6B). LFPs reflect population of neuronal activities around the recording electrode placed brain region [29]. LFPs were evoked in somato and thalamus region during sensory stimulation in sham-operated mice, whereas in PFC, LFPs were diminished during the stimulation (Figure 6C, left). These findings suggest that evoked LFPs in PFC show different properties from those of Somato and Thalamus.
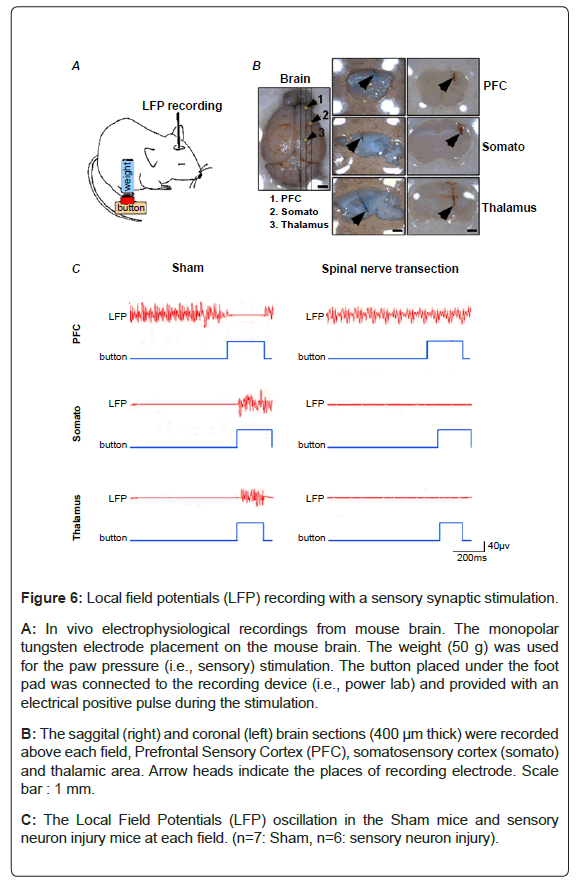
Figure 6: Local field potentials (LFP) recording with a sensory synaptic stimulation.
A: In vivo electrophysiological recordings from mouse brain. The monopolar tungsten electrode placement on the mouse brain. The weight (50 g) was used for the paw pressure (i.e., sensory) stimulation. The button placed under the foot pad was connected to the recording device (i.e., power lab) and provided with an electrical positive pulse during the stimulation.
B: The saggital (right) and coronal (left) brain sections (400 μm thick) were recorded above each field, Prefrontal Sensory Cortex (PFC), somatosensory cortex (somato) and thalamic area. Arrow heads indicate the places of recording electrode. Scale bar : 1 mm.
C: The Local Field Potentials (LFP) oscillation in the Sham mice and sensory neuron injury mice at each field. (n=7: Sham, n=6: sensory neuron injury).
Next, to examine whether the evoked LFPs are mediated by the peripheral nerve activation, the spinal nerve of mice were transected. In spinal nerve transected mice, the alteration of evoked LFPs during paw pressure stimuli were ablated (Figure 6C, right). These findings suggest that the evoked LFPs of the somato and thalamus are induced with peripheral nerve activation, and the activation of the somato and thalamus inhibits the PFC activation.
SE aggravates the evoked LFPs: To examine the neuronal dysfunction with SE, LFPs in PFC, Somato and Thalamus were recorded (Figure 7). In LPS and CLP-treated mice, the alteration of LFP amplitudes observed in sham-operated mice during the sensory stimulation (Figure 7B, left) was not found in each field (Figure 7B, middle and right). In addition, the results of the statistical analysis (Table 1) showed that the amplitudes of LFPs during stimulation were significantly increased in PFC, whereas decreased in somato and thalamus in the LPS and CLP-treated mice. Furthermore, all of these alterations were diminished by administration of GM6001. These findings suggest that the brain activities relating to somatosensory functions are aggravated via MMP-9 in SE.
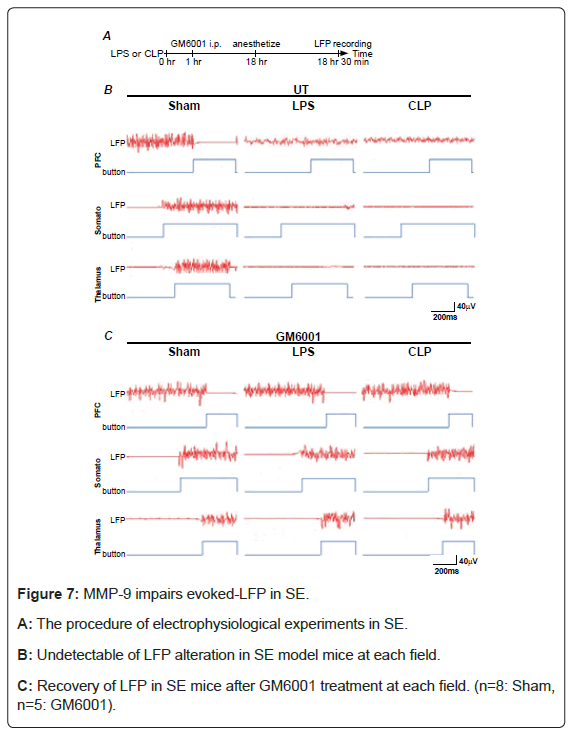
Figure 7: MMP-9 impairs evoked-LFP in SE.
A: The procedure of electrophysiological experiments in SE.
B: Undetectable of LFP alteration in SE model mice at each field.
C: Recovery of LFP in SE mice after GM6001 treatment at each field. (n=8: Sham, n=5: GM6001).
| PFC | Somato | Thalamus | ||
|---|---|---|---|---|
| UT | Sham | 0.07 ± 0.04 | 6.79 ± 0.35 | 8.45 ± 0.26 |
| LPS | 6.35 ± 0.18 | 0.99 ± 0.18 | 0.13 ± 0.08 | |
| CLP | 3.42 ± 0.43 | 0.25 ± 0.03 | 0.19 ± 0.02 | |
| Spinal Nerve transected | 9.25 ± 0.19 | 0.05 ± 0.02 | 0.03±0.01 | |
| GM6001 | Sham | 0.09 ± 0.05 | 6.85 ± 0.34 | 8.52 ± 0.24 |
| LPS | 0.06 ± 0.03 | 6.85 ± 0.39 | 8.39 ± 0.26 | |
| CLP | 0.07 ± 0.04 | 6.63 ± 0.35 | 8.23 ± 0.26 | |
Table 1: The amplitude (μV) of LFPs during stimulation.
In the present study, we found that increased MMP-9 caused gap junction degradation from the immunoblotting and immunohistochemical experiments. Several reports suggest that endotoxaemia induces the expression of MMP-9 [18,19,30]. These reports support our present finding that the activation of MMP-9 in cortical brain resulted in the synaptic dysfunction in SE.
What is the role of MMP-9? Conventionally, MMP-9 is well-known to cleave the extracellular matrix such as collagen, fibronectin etc, which allow migration of cancer cells for the metastasis [31] and of embryonic cells for the development [32]. However, recently, it is reported that MMP-9 affects the other substrates in the pathological conditions. For example, in the brain disorder, MMP-9 destroys the molecules such as tight junction protein composed of blood brain barrier and increases the permeability through the barrier [33]. In sepsis, MMP-9 aggravates septic condition with the promotion of tissue injury [34,35]. Hence, upregulated MMP-9 seems to cause the damages in the organs including lung, liver, kidney, spleen, stomach and brain accompanying with vascular hypofunction [18,19,35-38].
What is the detrimental effect of inducible MMP-9 to the brain dysfunction in SE? The MMP-9 expression is associated with the production of the pro-inflammatory cytokine such as IL-1β [39]. Our group previously reported that IL-1β from microglial cells results in synaptic plasticity deficiency in the hippocampus [17]. In addition, tumor necrosis factor-α [40,41] and C-X-C motif chemokine 10 (CXCL10) [42] which should be critical for the immune response in sepsis aggravated the normal brain function. These cytokines and inflammatory mediators from immune cells certainly lead to functional impairment of the brain in SE. Hence, MMP-9 not only degrades tight junction protein but also may trigger the pro-inflammatory cytokines which aggravate the brain dysfunction in SE. Furthermore, in the present study, we found a novel effect of MMP-9 on the reduction of connexin protein, which underlie the electrical synaptic transmission in the brain.
Connexin-43 is predominantly expressed on astrocyte in the brain (e.g. the frontal cortex, hippocampus and thalamus) [26,43,44]. The connexin supports neuron-astrocyte and astrocyte-astrocyte communications (i.e., transmissions of chemical substrates) which render a normal synaptic function [45]. In the present study, our findings clearly demonstrated that the increased active MMP-9 disrupts connexin-43, component of gap junction, which may lead to be the sensory neuronal dysfunction in SE. In addition, it was reported that the connexin-43 disruption may occur the increased gliosis and neuronal loss [43]. These neuronal dysfunctions reflect the reduction of LFP amplitudes during a sensory neuronal stimulation in SE (Figures 7A, 7B and Table 1). Hence, these pathological phenomena alter the neuronal activity in SE.
What is the patho-physiological dysfunction in SE? To tackle the issue, Local Field Potentials (LFPs) were recorded in thalamo-cortical network during the stimulation of sensory neurons. We found that LFPs in somatosensory cortex and thalamus evoked by sensory neuronal stimulation were lost in SE. These findings clearly demonstrated that the somatosensory dysfunction occurred in SE.
These findings are similar to LFP dissipation in PFC; however, the phenomena in the PFC seem to be unique (Figure 6). PFC is anatomically projected and functionally connected to somatosensory cortex and thalamus especially in pain stimulation [46]. However, in the PFC, brain activity alteration during somatosensory stimulation seems to be different from somatosensory cortex and thalamus [47]. Why is the somatosensory function related to PFC so unique? The answer may be found in the consistent activation of brain activity during the resting state, default mode. The default mode network is critical for the generation or maintenance of a normal brain function in the human mental state and behavior [48]. In a pathological condition, the aberrant default mode network is related to the unconscious state [49,50]. However, the patho-physiological mechanism is still unclear and warrant further evaluation.
Finally, MMP inhibitor, GM6001, clearly inhibits the active MMP-9 expression (Figure 1), reduction of occludin (Figure 2) and connexin-43 (Figure 3) and neuronal dysfunction (Figure 7) in SE. These findings suggest that active MMP-9 triggers the reduction of tight junction and gap junction proteins. Since GM6001 is, however, a broad spectrum inhibitor of MMPs, other candidate proteases (e.g. MMP-2) may be involved in these processes and warrant further evaluation. In summary, the regulation of MMP-9 activity is, at least, an important therapeutic target for the electrical synaptic dysfunction in SE.
This work was supported by grants from Grant-in Aid for Scientific research from the Japan Society for the Promotion of Sciences (24592734).
The authors declare no competing financial interests.