Journal of Clinical Chemistry and Laboratory Medicine
Open Access
ISSN: 2736-6588
ISSN: 2736-6588
Research Article - (2024)Volume 7, Issue 3
In this paper we have explored an ionic liquid as a matrix liquid to generate small molecule library on homogeneous phase soluble medium. In this context a rapid and effective method to prepare various isoindolo (2,l-a) benzimidazolone, pyrrolo (1,2-a) benzimidazolone and pyrido (1,2-a) benzimidazolone libraries on ionic liquid support by using focused microwave irradiation was described. The 4-fluoro-3-nitrobenzoic acid has been used as a building block to generate the benzimidazole heterocyclic core for library diversity through the ionic liquid supported multistep synthesis. Thereby various primary amines have been used to displace the fluorine atom through the nucleophilic substitution and subsequent reduction led to the IL supported conjugate diamine. The obtained conjugate diamne used as a scaffold to generate various ring size heterocyclic libraries via one pot intermolecular condensation with various substituent aliphatic γ and δ ketoacids in the presence of catalytic trifluoroaceticacid under microwave irradiation. This tandem operational simplicity procedure was extended to the aromatic γ and δ ketoacids, provided structurally heterogeneous libraries with skeletal diversity. The aforementioned three classes of heterocyclic lactam compounds further converted to thiolactams by using lawesson’s reagent. The relative high yield and purities with two points of diversity was reported.
Trifluoroaceticacid; Ketoacids; Heterocyclic libraries; Nucleophilic substitution; Intermolecular condensation
Structurally fused benzimidazole or quinazoline with an isoindolone, 2-pyrrolodinones and 2-pyridinone were embraced an important place in current therapeutic agents. Irrespective of the target, the best documented frequency in publications and the prescription drugs that containing the imidazole nuclei fused heterocycles have proven the remarkable ability of imidazole nuclei to serve both as biomimetics and reactive pharmacophores [1,2]. Thus, the Batracyclin (I) and the contracted 6 to 5 “C” ring of batracyclin known as isoindolo (2,1-a) benzimidazole (II) derivatives have showed in vivo antitumour activity against murine leukaemia P-388 and colon adenocarcinoma 38 cell lines [3]. They act as a topoisomerase II inhibitor and induce unscheduled DNA synthesis [4]. In continuous to that the related compounds of pyrrolo (l,2-a) imidazolones have showed as cognition enhancing pharmaceuticals [5]. In addition to this, the expanded series of pyrrolo (1,2-a) benzimidazolone (III) derivatives have been evaluated against the anticonvulsant properties, showed better activity [6]. Some of these scaffolds have been used in therapy, in particularly as nootropic agents [7]. The substituted Pyrimido (1,2-a) benzimidazole(IV) derivatives and the relevant anxiolytic agent RWJ-52763 analogues have been investigated as ligands for BZD site on GABA-A receptors are thus useful to treat the disorders of the central nervous system (Figure 1) [8].

Figure 1: Benzimidazole fused pharmacetically active heterocyclic molecules.
Enhancing the traditional paradigm of small molecule discovery, combinatorial chemistry has evolved in a dramatic increase in the number of compounds that are available for screening and to identify lead compounds for pharmaceutical research and drug development [9]. The increasing requirement of new compounds for drug discovery the fields of combinatorial and automated medicinal chemistry have emerged, where speed is of the essence [10]. Since a substantial effort has been dedicated to develop the chemistries, both solid and solution phase, for combinatorial syntheses of heterocyclic libraries [11]. The highly successful and readily automated insoluble solid resin support synthesis has been applied numerous areas [12]. However the problems associated with heterogeneous reaction conditions such as unequal distribution of the reaction sites and inefficient coupling rate has led to alternative soluble support methodologies with the aim to restore homogeneous reaction conditions. The successful employment of soluble Polyethylene Glycol (PEG), polyvinyl alcohol and other soluble polymers to synthesis oligopeptides, nucleotides, oligosaccharide and small molecule synthesis retained much advantage over conventional solution phase chemistry. However, the use of soluble polymer supports suffer from the drawbacks of low loading capacity, low aqueous solubility and energy intensive cooling required for purification [13-16]. The drawbacks associated with the solid phase and the soluble polymer supported phase synthesis to restore facile, convenient and nonpolluting synthetic procedures urges chemists to increase the tools are arsenal. The recently emerged Ionic Liquids (ILs) and their intriguing properties as environmentally benign reaction media have considerable interest in numerous chemical reactions and biomedical applications [17]. Depending on the choice of anions and cations the solubility of the ionic liquids can be tuned readily, thus allows the phase separation from organic as well as aqueous phase. These attractive features lend it to use the ionic liquids as soluble supports for organic synthesis [18]. Moreover, the substrate anchored on ionic liquids is allowed to monitor the conventional spectroscopic analysis (1H NMR, 13C NMR) during the synthetic process and the reactivity of supported substrate functional groups are expected to retain their reactivity as in solution phase. This liquid phase strategy has been demonstrated by several groups to synthesize small molecules and peptides in combinatorial fashion [19]. In each case the reaction conducted in homogeneous phase, the excess reagents and byproducts removed by simple washing with low polar organic solvent. The recovered ionic liquid, which was detached from the final product, can be reused to carry out the cycle of synthesis with loss of reactivity.
Synthetic organic chemistry together with the parallel synthesis significantly has much impacted by the introduction of precision controlled microwave reactors [20]. Through the application of microwave irradiation to parallel synthesis, the time frame for hit-to-lead optimization as well as the overall drug development process time can be significantly reduced. Ionic liquids absorb the microwave energy efficiently by which the reaction rate could be accelerated remarkably. The synergies arising from the combined use of MW and ionic liquid with tandem reaction procedures will certainly go a long way to meet the increasing demand for environmentally benign chemical processes. In this paper we described the advantages of ionic liquid supported synthesis from the selection of non-limiting examples of ionic liquids together with microwave heating as a greener alternative in the synthesis of bioactive heterocyclic compounds. To illustrate the rapid and efficient synthetic methodology, to generate aforementioned three classes of heterocycles with two points of skeletal diversity, on rapid time scale with high yield and relative purities are summarized.
The general concept of ionic liquid supported synthesis in accordance with the present synthetic library has been illustrates in Figures 1 and 2. The 3-hydroxyethyl-(1-methylimidazolium)- tetrafluoroborate readily available from the reaction of 1- methylimidazole and 2-bromoethanol was chosen as a nonlimiting example of a suitable ionic liquid support to illustrate the ionic liquid supported synthesis from the published literature. The 4-fluoro-3-nitrobenzoicacid has been used as a building block in order to create the benzimidazole fused chemotype molecules through the tandem annulation process with various ranges of ketoacids. Synthetic strategy in this developing distinct molecular framework structures, 4-fluoro-3- nitrobenzoicacid was attached to the ionic liquid through the ester bond where IL acts as a soluble support anchor to the growing molecules (Table 1). This reaction has been carried out in the presence of DCC and the catalytic amount of DMAP in acetonitrile and dichloromethane as a co-solvent for about 20 minutes in the microwave irradiaton to offered IL conjugate. Since the ionic nature of ILs, the efficient absorption of microwave irradiation can increased the rate of the reaction even for low polarity reaction media. In order to offer the product analogues under conventional heating methods required 2 days for the completion of reaction. By taking the advantage of distinct solubility feature of the IL anchored substrate, the excess reagents and the reaction byproducts have been removed using less polar organic solvents in which the IL conjugate substrate insoluble. The ionic liquid supported 4-fluoro-3-nitrobenzene was treated with various primary amines to replace the fluorine atom through ipsofluoro nucleophilic substitution reactions to generate structurally distinct frameworks under focused microwave irradiation for about 10 min to get the ionic liquid supported conjugate. On the other hand this reaction took place for about 10 h under reflux conditions. In order to reduce nitro functional group, the IL anchored substrate treated with zinc and ammonium formate for about 20 minutes at the room temperature to furnish IL conjugate diamine.

Figure 2: Ionic liquid supported synthesis of pyrrolobenzimidazolone, and pyrido-benzimidazolone derivatives.
 |
||||
|---|---|---|---|---|
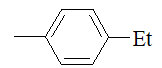
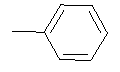
| Entry | NH2-R1 | R2 | R3 | n/X |
| 10a | 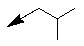 |
-CH3 | -H | 1/0 |
| 10b |  |
-CH3 | -H | 1/0 |
| 10c | 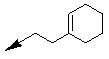 |
-CH3 | -H | 1/0 |
| 10d | 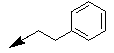 |
-CH3 | -H | 1/0 |
| 10e | 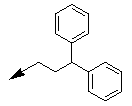 |
-CH3 | -H | 1/0 |
| 10f | 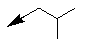 |
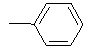 |
-H | 1/0 |
| 10g | 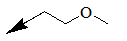 |
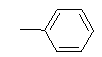 |
H | 1/0 |
| 10h | 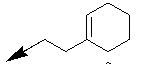 |
 |
-H | 1/0 |
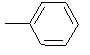
| 10i | 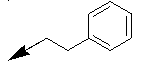 |
 |
-H | 1/0 |
| 10j | 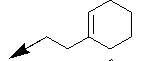 |
 |
-CH3 | 1/0 |
| 10k | 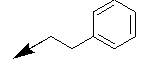 |
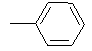 |
-CH3 | 1/0 |
| 10l | 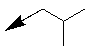 |
-CH3 | -H | 2/0 |
| 10m | 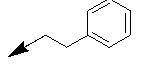 |
-CH3 | -H | 2/0 |
| 10n |  |
-CH3 | -H | 2/0 |
| 10o |  |
-CH3 | -H | 2/0 |
| 10p |  |
-CH3 | -H | 2/0 |
| 10q | 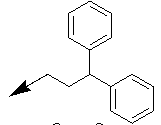 |
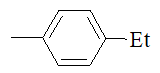 |
-H | 1/0 |
| 10r |  |
 |
-H | 1/0 |
| 10s |  |
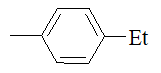 |
-H | 1/0 |
| 10t |  |
 |
-H | 1/0 |
| 10u | 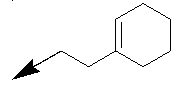 |
 |
-H | 2/0 |
| 10v | 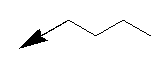 |
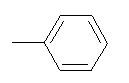 |
-H | 2/0 |
| 10x | 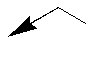 |
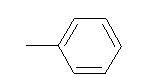 |
-H | 2/0 |
| 11a |  |
-CH3 | -H | 1/S |
| 11b |  |
 |
-H | 1/S |
| 11c |  |
-CH3 | -CH3 | 1/S |
| 11d |  |
-CH3 | -H | 2/S |
| 11e |  |
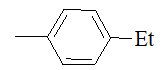 |
-H | 1/S |
| 11f | 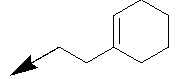 |
 |
-H | 2/S |
Table 1: Synthesis of pyrrolo, pyrido-benzimidazolones with two different appendages.
Further the diamine has been used to treat various aliphatic γ and δ ketoacids 6 to produce structurally diverse heterocyclic libraries. This reaction has been carried out by using catalytic amount trifluoroacetic acid and anhydrous magnesium sulfate under focused microwave irradiation for about 10 minutes at 110℃ to offered 8 whereas the to furnish the same reaction under reflux conditions took 24 h. During the optimization of the reaction conditions we have been observed 7 was the product upon the addition of 5% of trifluoroaceticacid in ethlenedichloride under focused microwave irradiation. The formation of the β keto tethered imidazole 7 nucleuses and their represented structure have much bioactivity impact (Figure 3 and Table 2).

Figure 3: Synthesis of isoindolo-benzimidazole derivatives.
| Entry | NH2--R1 | R2 | X |
|---|---|---|---|
| 16a |  |
-CH3 | O |
| 16b |  |
-CH3 | O |
| 16c | 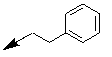 |
-CH3 | O |
| 16d |  |
-CH3 | O |
| 16e | 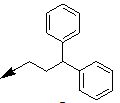 |
-CH3 | O |
| 17a | 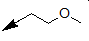 |
-CH3 | S |
Table 2: Synthesis of isoindolo-benzimidazolones with two different appendages.
To extend the generality of this methodology ionic liquid conjugated diamine further treated with various aromatic γ ketoacid 12 using catalytic trifluoroaceticacid and the MgSO4 as dehydrating agent under microwave irradiation at 110℃ for about 10 min reaction time offered structurally distinct library of analogues. Whereas the same reaction by the addition of 5% of trifluoroaceticacid under microwave irradiation at 110℃ the formation keto tethered benzimidazole has been observed as a side product. The mechanistic consideration of the formation of the products under focused microwave condition and catalytic amount of trifluoroaceticacid was understood that the acid catalyzed emine formation by the expense of liberating water molecule, thereby subsequent secondary amine attack on the electrophilic imine carbon leads further amide bond formation in one pot cyclic five and six membered imidazole fused cyclic amides. The formation tandem one pot transformation from tethered ketoacid by the expense of liberating water molecule was absorbed by the dehydrating agent magnesium sulphate. The further thionation of the amide functional group target library compounds 8 and 14 has been carried out by using Lawesson’s reagent under microwave irradiation at 150℃ for about 15 min to furnish. It is noteworthy to mention that the time taken to attain the desired reaction temperatures for all the reactions under microwave exposure has substantially shorted period of time scale, because of the high polar microwave absorbance medium created by the ionic liquid support otherwise it might be took longer reaction times. The process of thionation of the library has been carried out in toluene at 150℃, the reaction temperature attained in the short period of times in the presence of ionic liquid support even in the less polar solvents. Moreover, the solubility of the ionic liquid support throughout the sequence of reaction was not been influenced by the growing chain. Thus allows the separation and purification procedures become greatly simplified the general procedure involved washing the steps with aqueous and organic solvents. The reported synthesis of 1H-imidazo (2,1-a) isoindole-2,5 (3H, 9bH)-diones and related pyrrolo (1,2-a) benzimidazol-1-one, and pyrido (1,2-a) benzimidazol-1 (2H)-one derivatives are involved in time consuming and tedious process, afford moderate overall yields, and the R1 group is limited in published examples to hydrogen. The lack of appropriate positional diversity around the benzimidazole scaffold requires significant effort to optimize the synthetic procedure by which novel compounds can be prepared. We have been reported here soluble support together with the microwave irradiation on rapid phase synthesis with two points of diversity to generate various heterocyclic molecules with high purity and yields over the tedious and time consuming process. The reaction progression was monitored through the proton NMR spectrum without release from the support, the formation of the analogues can figure out that 6.8 ppm doublet is the characteristic peak for the characteristic peak of aromatic proton and also we can figure out that the methyl group protons at the 1.7 ppm singlet. While the formation of the keto tethered benzimidazole analogues 7 and 13 derivatives have been figure out by changing the variation of characteristic peaks from spectrums where the aromatic and the methyl proton has been shifted to the downfield 7.2 ppm and 2.0 ppm respectively. The formation of the stereogenic quaternary carbon center and the amide bond has been found out from the 13C NMR at 83.0 ppm-89.5 ppm along with the amide bond at 170.5 ppm. Further the formation of amide bond has been confirmed by the IR spectroscopy. Finally, the products those cleaved from the Ionic liquid support has been carried out by using NaOMe in methanol under microwave irradiation at 110℃ for about 10 min to furnish. The cleaved products have been extracted from the extraction by using dichloromethane (3 ml × 50 ml). The crude final compounds subjected to HPLC purity and the recovered Ionic liquid has been used to cycle the synthetic pathway without loss of activity.
Experimental section
General methods: Microwave assisted Ionic liquid supported synthesis here in we described throughout the manuscript have been performed under CEM discover microwave system at the frequency of 2450 Hz (0 W-300 W) in closed vessel system. Dichloromethane was distilled from calcium hydride before use. All reactions were performed under an inert atmosphere with unpurified reagents and dry solvents. Analytical Thin-Layer Chromatography (TLC) was performed using 0.25 mm silica gelcoated Kieselgel 60 F254 plates. Flash chromatography was performed using the indicated solvent and silica gel 60 (Merck, 230 mesh-400 mesh). All the microwave experiments were conducted in a CEM discover microwave system under optimized conditions of power and pressure. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Bruker DX-300 spectrometer. Chemical shifts are reported in parts per million (ppm) on the δ scale from an internal standard. High-Resolution Mass Spectra (HRMS) were recorded on a JEOL TMS-HX 110 mass spectrometer.
General procedure for the synthesis of the pyrrolo, pyridobenzimidazolones and isoindolo-benzimidazolones derivatives
Ionic Liquid 1 (0.5 g, 0.2 mmol) in acetonitrile (5 mL) was added to the solution of 4-fluro-3-nitrobenzoic acid 2 (0.51 g, 2.7 mmol, 1.2 equiv) in dichloromethane/acetonitrile (5 mL) in the presence of N,N’–dicyclohexylcarbodiimide (0.57 g, 2.7 mmol, 1.2 equiv) as coupling reagent and a catalytic amount of N,N’- dimethylamino pyridine (DMAP, 0.002 g) under microwave irradiation at 80℃ for about 20 minutes to afford the ester 3. The precipitated Dicyclohexyl Urea (DCU) was filtered through a fritted celite plug. The celite plug was rinsed with acetinitrile and the combined organic phase was concentrated under vacuum. The crude reaction mixtures were precipitated by slow addition of excess of cold ether (100 mL). The precipitated ester conjugate was then filtered through a fritted funnel and washed several times to remove the byproducts and dried. To a solution of 3 in acetonitrile (5 mL), various primary amines (6.7 mmol, 3.0 equiv) in (5 mL) dichloromethane was added and the reaction mixtures were irradiated in microwave cavity at 80℃for 10 min to afford the ionic-liquid immobilized nitroamines. Upon concentration under reduced pressure, the crude residue was precipitated by addition of excess of cold ether (100 mL). The precipitated IL conjugate was then filtered through a fritted funnel and washed several times to remove the byproducts and dried. The subsequent reduction of nitro group proceeded by using Zn (7.0 equiv) and NH4COOH (15.0 equiv) in methanol at room temperature for about 15 minutes under vigorous stirring. The insoluble Zn was removed by filtration and the mixture was dried under vacuum. The obtained residue was dissolve in dichloromethane and washed with 2M HCI (10 mL × 3). The organic phase was dried over MgSO4 and concentrated to afford ionic liquid supported dimaine. To the solution of 5 in ethylenedichloride (9 mL), various aliphatic γ or δ ketoacids (1.5 equiv) were added in the presence of dry MgSO4 or 4 Ǻ molecular sieves and the catalytic amount of trifluoroaceticacid, subjected to microwave irradiated under microwave for about 10 minutes at 110℃ to offered the IL supported conjugates. To the solution of same 5 in ethylenedichloride (9 mL) with various aromatic γ or δ ketoacids (1.5 equiv) were added in the presence of dry MgSO4 or 4 Ǻ molecular sieves and the catalytic amount of trifluoroaceticacid, subjected to microwave irradiated under microwave for about 10 minutes at 110℃ to offered the IL supported conjugates. The crude reaction mixture was filtered through the fritted funnel and the reaction mixture was precipitated by slow addition of excess of cold ether (60 mL). The precipitated Ionic liquid conjugate was then filtered through a fritted funnel and washed several times to remove the byproducts. Thereby the fritted funnel was rinsed with dichloroethane and the combined organic phase was concentrated under vacuum. To the solution of IL supported conjugates in toluene (10 mL), was added by the Lowesson’s reagent (0.7 equiv) and subjected to MW irradiation for about 10 min at 170℃, to offered derivatives. The solvent evacuated crude residue has been precipitated by the addition of excess cold ether (100 mL). The precipitation of IL conjugate filtered through the fritted funnel, fritted funnel was rinsed acetonitrile and thereby evaporated the solvent. The solution of IL support conjugates were added by 0.1 M NaOMe in MeOH (10 mL) and subjected to MW irradiation for about 10 min at 110℃ to cleave the IL support. The solvent evacuated crude residue was extracted from dichloromethane (3 mL × 10 mL). The purity of the obtained crude compounds has been evaluated by the HPLC, subsequent column chromatography over silica gel using ethylacetate/n-hexane (1:1) as eluent to obtain the derivatives.
In summary, we have developed rapid and efficient solution phase approach to synthesize isoindolo (2,l-a) benzimidazole, related pyrrolo (1,2-a) Benzimidazol-1-one, and pyrido (1,2-a) benzimidazol-1(2H)-one along with their related thionated derivatives in high yield and purities. Combining these ionic liquid supported tandem synthesis together with the microwave heating conceptual advances embodies tandem synthesis is a powerful new approach to synthesize parallel libraries on rapid time scale have been successfully accomplished. The employed IL support strategy offers several advantages over the solid and liquid phase organic synthesis. The progression of reaction intermediates could be monitored by the conventional spectroscopic methods like 1H, 13C, MS. This effective methodology in associated with high loading capacity and commercial availability considerations expands the strategies for synthesis of such compounds and permits much larger number of these ‘drug-like’ molecules to be prepared.
Financial support from the National Science Council (NSC) of Taiwan and the authorities of the national Chiao Tung university, for providing laboratory facilities are gratefully acknowledged.
The detailed experimental procedure for the synthesis of compounds; spectral characteristics of the synthesized compounds 1H NMR, 13C NMR and chiral HPLC spectra of compounds were available.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Thummanagoti S (2024) Microwave Assisted Tandem Transformation Led to Pyrrolo, Pyrido-Benzimidazolones and Isoindolo- Benzimidazolones on Ionic Liquid Support. J Clin Chem Lab Med. 6:294.
Received: 15-Mar-2023, Manuscript No. JCCLM-23-22174; Editor assigned: 17-Mar-2023, Pre QC No. JCCLM-23-22174 (PQ); Reviewed: 31-Mar-2023, QC No. JCCLM-23-22174; Revised: 18-May-2023, Manuscript No. JCCLM-23-22174 (R); Published: 25-Sep-2024 , DOI: 10.35248/2736-6588.24.7.294
Copyright: © 2024 Thummanagoti S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited