Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2013) Volume 2, Issue 1
Neurodegeneration and cancer are fast becoming the leading causes of age-associated disability, dementia and ultimately death worldwide. Although oxidative stress has been intensely studied, little analysis has been done in chronic oxidative stress-induced mitochondrial models. In this regard, DNA-overproliferation and/or deletion initiate mitochondrial deregulation causing energy failure, which has been implicated in the pathogenesis of Alzheimer’s disease (AD), tumor growth, and metastasis. In this regard, decline in mitochondrial normal homeostasis during the development and maturation of the neurodegeneration, tumor growth, and/or metastases is characterized by tissue and cellular oxygen deficiency, which leads to subcellular energy defects. In addition, the overexpression of the cascades initiates the formation and release of large amounts of reactive free radicals [mainly nitric oxide (NO)
via the overexpression of NO synthases], which cause the oxidative stress, cellular alterations, and concomitant mitochondrial lesions and decline in normal organ function. The present study explores the intimate, i.e. direct relationship between chronic oxidative stress and mitochondrial damage as a vital life-supporter for cells and/or the microcirculatory systems whose damage occurs before the development of human AD. Our study highlights the effects of chronic oxidative stress-induced mitochondrial DNA over-proliferation and/or deletion and mitochondrial enzyme activities during the development of human AD. Mitochondrial DNA damage also leads to other pathologies, including colorectal cancer in liver metastasis, and malignant brain cancers. We hypothesize that mitochondrial lesions, especially mitochondrial DNA abnormalities, are detrimental to cell viability and thus mitochondrial DNA damage could be used as a new diagnostic tool and/or criterion for the early detection of AD and other diseases. Further extension of this approach will enable us not only for the better understanding of the blood brain barrier (BBB) homeostasis, which most likely plays a key role in the development of AD and some of forms of the cancer, but also for the development of new and more specific treatment strategies.
Keywords: Oxidative stress, Mitochondria, DNA deletions, Alzheimer disease, Cancer, Antioxidants, Drug development
The growing body of different experimental models that mimics AD and cancer, as well as clinical observations, strongly suggests that chronic hypoperfusion contributes to the pathogenesis of neurodegeneration and cancer development [1-7]. Hypoperfusion appears to induce oxidative stress characterized by increased levels of oxidative stress markers such as overproduction of NO, and superoxide dismutase [2,8]. Oxidative imbalance is also known to correlate with the onset or complications of neurodegenerative disease, including AD and other age-related diseases such as atherosclerosis and rheumatic arthritis and diabetes mellitus [9].
We have demonstrated that the sustained hypoperfusion induced by continues chronic injury stimulus results in energy failure due to mitochondrial ultrastructure damage, leading to an overproduction and/or deletion of mitochondrial DNA (mtDNA) [7,10-12]. Concomitant to mitochondrial abnormalities lipid peroxidation also produces a variety of nucleic acid-damaging agents [7,10-12]. The oxidation event occurs mainly within the vascular endothelium, where it causes structural changes in the mitochondria [10]. Moreover, these pathological events are always accompanied by neuronal and glial cells damages, considered being integral in the development of AD and/or AD-like pathology [10,11]. Moreover, we have found that the microvascular lesions in the AD brain are positively and linearly correlated with the severity and degree of neuronal and glial cell lesions in these regions. Because the brain exhibits high energy demand, coincidental generation of reactive oxygen species/reactive nitrogen species (ROS/RNS) and vulnerability to oxidative stress occurs, thus central substrate of brain lesions in AD has been revealed to be caused by mitochondrial damage comprising of DNA deletions and overexpression of oxidative stress markers in cellular compartments [7,10-12]. This eventually halts energy production and effectuates cognitive impairment and memory decline.
Age-associated brain microvascular hypoperfusion is a primary pathogenic factor for the onset of AD and related pathologies [10,11,13,14]. Moderate and/or severe cognitive clinical sign is often accompanied by presence of vascular risk factors and cerebral hypoperfusion often coexists with cerebral vascular oxidative imbalance [15-17]. Ultrastructural features of vascular wall cells in human AD are also common to the yeast artificial chromosome (YAC R140) and C57B6/SJL transgenic (Tg+) mice overexpressing amyloid beta precursor protein (AβPP). However, the relevant age-matched controls of human AD, YAC and C57B6/SJL Tg(+) mice exhibit nonconsiderable vascular amyloid deposition in contrast to the diseased brain models [10,11]. When probed with wild type and deletedmtDNA probes, microvessels from undamaged human AD, YAC and C57B6/SJL transgenic (Tg+) mouse tissues show a complete lack of any abnormalities unlike the cell patches positive for deleted mtDNA in the study models that mimics human AD and human AD brain tissues [10,11]. These observations indicate that the oxidative stress induces continues damage on vascular wall cells, especially on their mitochondria, preceding the development of AD pathology [10,11,18].
ROS and RNS are continuously generated at sites of injury and/ or inflammation. Low concentrations of ROS and RNS are required for various physiological activities including signaling and apoptosis [11,19-21], but at higher concentrations these free radicals induce cellular dysfunction, injury and, ultimately with cell-death. The vascular endothelial cells (EC), which regulates the vascular tone, circulation of nutrients between intra- and extravascular space and releasing vasoactive substances including neuropeptides that controls blood flow by modulating the coagulation is a major target of oxidant stress and their products [18]. This tiny cellular layer play a crucial role in the organ and tissues homeostasis and appeared to be primary targets in the pathophysiology of vascular diseases, autoimmune disorders, inflammation and any diseases including tumor growth and metastases [11,19-21]. In addition, the vascular endothelium, neurons, and glial are both the source and target of ROS/RNS, and respond to certain stimuli, especially to chronic hypoxia/hypoperfusion where the endothelial, neuronal and glial intracellular ROS production significantly increased. Cellular hypoperfusion is a prominent inducer of vascular lesions and has been experimentally indicated to be the initiator of AD [22,18]. We hypothesize that the hypoperfusion-induced accumulation of ROS and RNS results in the pathogenesis of AD by promoting leukocyte adhesion and altering normal vascular function with the end result of organ or tissue dysfunction [23].
Oxidative stress stimulates a secondary overexpression of iNOS and nNOS and endothelin-1 (ET-1), especially in neuronal cells involving high metabolic activity especially on highly metabolic activity cells such as neurons [13]. Moreover, features of the neural tissue is extremely prone to oxidative damage because of the (a) high rate of oxygen consumption, (b) high polyunsaturated lipid content, (c) low levels of classic antioxidant enzymes [23]. One of the key features of the AD brain appeared to be overexpression of oxidative stress markers [24-26], which show evidence of decline in polyunsaturated fatty acid [27,28]; elevation in LPO [29,30]; oxidation of biomolecules protein [31,32], DNA [11,19-21], RNA [33-35] and are accompanied in AD brains. Further, various advanced glycation end products (AGEs) such as trans-4-hydroxy-2-nonenal (HNE) and Nε-carboxymethyllysine (CML) have been observed in both neurofibrillary tangles (NFT) and senile plaques (SP) in AD brain [15,24,25] as well as in post–ischemic tissues [36-38]. It is therefore reasonable to assume that overexpression of oxidative stress markers contribute to brain parenchymal cell damage and dysfunction of the BBB, which is critical in regulating the microenvironment of the brain, optimizing neuronal function.
Defective Aβ clearance may also contribute to the disease pathology. Aβ clearance across the BBB is receptor mediated (mediated by RAGE, receptor for advanced glycation end products and LPR, lipoprotein receptor-related protein receptors) occurring via perivascular macrophages, perivasculature and the microglia compartment [39-41]. Decreased clearance of Aβ across the BBB is associated with formation of neuritic plaques which are cleared upon Aβ antibody therapy in AD patients and mouse models that mimics this pathology [15,40,42].
The highly dynamic mitochondrion is the main supplier of energy to the cell and the cellular homeostasis regulators. It has been demonstrated that the mitochondria drive adenosine three phosphate (ATP) synthesis via the electron transport chain through the synergistic actions of complex I, III, IV (cytochrome c oxidase, COX) and ATP synthase [43]. Alzheimer pathology co-occurs with declined mitochondrial bioenergetics potential, impaired oxidative phosphorylation and the resultant overt free radical production and oxidative damage [2,3,12,15,44,41]. Thus, the oxidative stress plays a major role in the neurodegenerative diseases, in particular AD [2,3,45-47]. Following long-term ischemia/reperfusion results in the disintegration of mitochondrial ultrastructure, as demonstrated by in vivo and in vitro models [15,44,46,47]. Yao et al. [12] experimentally demonstrated that dysregulation of mitochondrial bioenergetic mechanisms are associated with AD pathogenesis. They showed that in female triple transgenic AD mouse model (3xTg-AD) decreased mitochondrial respiration, increased hydrogen peroxide (H2O2) production and lipid peroxidation preceded the development of observable plaque formation. Furthermore, reproductive senescence age aggravated mitochondrial dysfunction in conjunction with increase in AD pathology. Thus indicating that mitochondrial dysfunction occurs early in AD pathogenesis [2,11,12,15,44-46,48-55]. The most noted deregulation in mitochondrial enzyme activity is the reduction of COX activity in different brain regions and cellular compartment of AD patients having serious consequences on energy metabolism [3,15,33-35,56-58]. Effects of other mitochondrial oxidative phosphorylation complexes are less documented. Certain mutations affecting the L-strand transcription effectuating inhibition of complex I respiration, increase in ROS production, decline in membrane potential and deregulation of calcium homeostasis have been related to opening of mitochondrial permeability transition pore (mPTP) and neuronal death [43]. Aβ mediated oxidative damage and direct inhibition of mitochondrial enzymes also contributes substantially to AD pathology. Aβ directly inhibits COX [59], interacts with cyclophilin D [60], oxidatively damages Drp1 and is correlated with altered levels of mitochondrial fission/fusion proteins [61,62]. Aβ mediates mitochondrial failure by yet another protein-protein interaction. It binds to Aβ binding alcohol dehydrogenase (ABAD) a matrix protein and prevents the binding of NAD+ to the enzyme. This interaction results in increased ROS production, change in mitochondrial membrane permeability, cell death and cognitive impairment in APP/ABAD transgenic mouse model [63-65]. End result is increased mitochondrial damage and fission and decline in mtDNA copy number. Impairment of mitochondrial function initiates a vicious cycle of enhanced ROS production, further damage to mitochondrial enzymes and mtDNA, and Aβ formation. These events in feed forward mechanism worsen the AD pathology.
Studies using in situ markers for 8-hydroxydeoxyguanosine (8- OHdG) and 8-hydroxyguanosine (8-OHG) showed that RNA oxidation is also a prominent feature of damaged neurons in AD [3,15,33-35]. Yao et al. [12] have suggested that if mitochondrial dysfunction endangers AD then environmental and genetic factors to which mitochondria are susceptible must also be critical for the development of late onset sporadic AD. Offspring with maternal history of AD show a relationship between brain hypometabolism and increased risk of AD [66]. Maternal inheritance of mitochondrial genome supports Yao et al. [12], providing evidence of mitochondrial dysfunction as a causative factor in AD pathogenesis.
Our ongoing studies by using different techniques demonstrated that the mitochondrial lesions appear to be primary hallmark of the colorectal liver tumors derived from colon as well as primary brain tumor such as glioblastoma [48]. Vessel endothelium from microcirculatory systems of the tumor tissues shows the damage of mitochondrial cristae. In addition, the mitochondria derived lysosomal and vacuolar structures appear to be a permanent feature of the glioma derived tumor cells. Especially, the lipid laden tumor cells and surrounding cells often display varying degrees of mitochondrial abnormalities (such as mitochondria with broken cristae, presence of edema in their matrix, disruption of inner and external mitochondrial membrane). Moreover, giant mitochondria also are permanent features of tumor growth and metastases [6,67]. The marginal and central portion of tumor tissues obtained from patients undergoing surgery of the primary glioblastoma show heterogeneous lesions in the structure of the mitochondria. In particular, in the central regions of tumor tissues, astrocytes are associated with clusters of mitochondria derived lysosomes [67]. The same patterns of cellular and subcellular damage were also seen in spinal cord tumor [67].
The mitochondrial DNA overproliferation and deletion was detected by using cytological in situ hybridization in tumor cells and AD tissues [11,68]. Our study demonstrated that successful dysregulation of cell cycle, and that early cell-cycle pathophysiology in AD may recruit oncogenic signal transduction mechanisms, which we hypothesized as an abortive neoplastic transformation that appeared to be prominent during tumorigenesis and AD [69]. We were able to demonstrate that abnormal mitochondria and lipofuscinogenesis appeared as a feature of hippocampal neuronal and other cellular damage during the development and maturation of human AD and AD mice that mimics AD-like pathology, and suggest a direct relationship between vascular abnormalities, BBB breakdown, neuronal loss and amyloid depositions [11,68,70]. However, such abnormalities are characterized by a heterogeneous distribution, including the presence of a giant and electron dense mitochondria that can be seen in different population of the brain cells including hippocampal neurons [11,68]. In situ hybridization analysis showed that the majority of human and mouse specific mtDNA deletions were found in mitochondrial-derived lysosomes in the neuronal cell body which are closely associated with lipofuscin. These results indicates that proliferation, deletion and duplication of mtDNA occurs in mitochondria, many of which have been fused with lysosomes in human AD [70-73], and transgenic mice as a model for neurodegeneration [70-73]. The common features which we saw on the mitochondrial abnormality in the brain during the tumorigenesis and in AD, strongly suggesting that most likely mitochondrial DNA overproliferation/deletion appeared to be key initiating factors for tumor growth/metastases whose similarity is seen in AD [11,68,72,73]. We hypothesize that the investigating mitochondrial abnormality is most likely able to open new windows not only to better understand tumor pathogenesis but also new and more effective and specific treatment strategies in these conditions.
Damaged mitochondria is heavily involved in the generation of ROS and results in oxidative damage to the vascular endothelium, as well as to other cellular constitutes in tumor tissues that initiate complication and induces tumor pathology. Brain disorders that involve chronic hypoperfusion may be responsible for concomitant energy failure and the pathogenesis that underlies both disease processes, as hypoperfusion appears to induce oxidative stress, which is largely from ROS as well as NO [3,5,7,13,14].
An increase in the release of NO from the tumor tissue [vascular endothelium and other tumor tissue cells, the natural killer cells (NKHlymphocytes)], is able to promote anti-tumor growth activity, kill cancer cells and can be utilized for eliminating free radicals, which are increased in tumor growth and metastatic conditions. We demonstrated that the metastatic colorectal cancer to liver and malignant brain cancer are characterized by overexpression of all 3 of NOS enzymes isoforms, which coexist with mitochondrial ultrastructural alterations in tumor cells. Moreover, the severity and degree of tumor growth and metastasis are linearly correlated with the overexpression of iNOS and increased level of ET-1 immunoreactivity [74]. The role of ET-1 as a mitogen in the pathogenesis of tumor growth and metastasis has been studied extensively by our group and other research team [6,67,75]. We showed that the expression of ET-1 immunoreactivity not only in vascular endothelium, but also in tumor cells, activated lymphocytes, SMC, and in liver hepatocytes [6,67,76]. We speculate that most likely this positive feedback appears to be a compensatory action of tumor invaded organs during the tumor growth and metastases. However, exact cellular mechanisms of tumor vascular growth and its dependence on the oxidative stress markers such as nitrotyrosine, lipid peroxidation, mtDNA deletion is not clearly understood [6,67,76]. Further studies on mitochondrial damage due to NO mediated oxidative stress are needed to address this issue [11,68,72]. Of particular importance, these studies are expected to lead to new avenue to not only to better understand the pathophysiology but also to provide more effective treatment strategies for cancer [67].
Ultrastructural investigation of the brain microcirculatory systems from age-matched control (Figure 1A) and AD (Figures 1B-D) patients are characterized by heterogeneous morphology. The short postmortem (<2 h) brain tissues from AD patients, displays microvessels with different degree of lesions, characterized by the presence of clusters of nonreversible damaged mitochondria (mitochondria without any cristae etc.) and/or mitochondria derived lysosomes in these microvessels endothelium (Figure 1B).
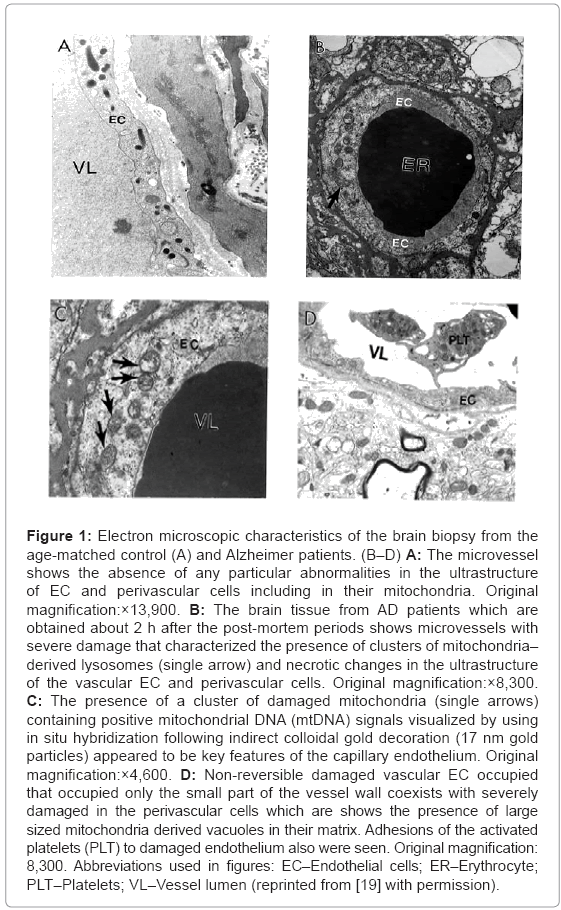
Figure 1: Electron microscopic characteristics of the brain biopsy from the age-matched control (A) and Alzheimer patients. (B–D) A: The microvessel shows the absence of any particular abnormalities in the ultrastructure of EC and perivascular cells including in their mitochondria. Original magnification:×13,900. B: The brain tissue from AD patients which are obtained about 2 h after the post-mortem periods shows microvessels with severe damage that characterized the presence of clusters of mitochondria– derived lysosomes (single arrow) and necrotic changes in the ultrastructure of the vascular EC and perivascular cells. Original magnification:×8,300. C: The presence of a cluster of damaged mitochondria (single arrows) containing positive mitochondrial DNA (mtDNA) signals visualized by using in situ hybridization following indirect colloidal gold decoration (17 nm gold particles) appeared to be key features of the capillary endothelium. Original magnification:×4,600. D: Non-reversible damaged vascular EC occupied that occupied only the small part of the vessel wall coexists with severely damaged in the perivascular cells which are shows the presence of large sized mitochondria derived vacuoles in their matrix. Adhesions of the activated platelets (PLT) to damaged endothelium also were seen. Original magnification: 8,300. Abbreviations used in figures: EC–Endothelial cells; ER–Erythrocyte; PLT–Platelets; VL–Vessel lumen (reprinted from [19] with permission).
In situ hybridization following indirect 17 nanometer colloidal gold decoration shows the presence of a cluster of damaged mitochondria with positive mtDNA signals (Figure 1C). In AD brain microvessels EC occupied a fraction of the vessel wall, and the adhesion of the activated platelets (PLT) to damaged endothelium appears to be a hallmark of the damaged microvessels (Figure 1D). Undamaged vessel endothelium, however, showed no ultrastructural changes, and the mitochondria appear to be intact (Figure 2A). Microvessels in the perivascular spaces contain large vacuolar structures (Figure 2) and EC show the presence of degenerative mitochondria (Figure 2B). Electron-dense hypoxic mitochondria coexist with the mitochondria derived lysosomal structure in the cytoplasmic matrix of EC and perivascular cells (Figure 2C). Hypoxic and/or completely damaged mitochondria exhibit excessive endothelium and perivascular cell damage (Figure 2D).

Figure 2: Ultrastructural features of brain microvessels from AD brains. A: The microvessel endothelium with the intact morphology did not show any particular changes in their ultrastructure (single arrows indicate intact mitochondria). However, the perivascular cells show the presence of large vacuolar structures in their matrix (double arrow). Original magnification× 13,000. B: EC shows the presence of degenerative mitochondria (double arrow). Original magnification×6,600. C: The electron-dense hypoxic mitochondrion (single arrows) coexists with the formation of mitochondria derived secondary lysosomal structure in the cytoplasmic matrix of EC and perivascular cells (indicated by double arrow). Original magnification×20,000. D: Mitochondria abnormality coexists hallmark of the vascular endothelium and perivascular cells (single and double arrows indicate hypoxic and completely damaged mitochondria, respectively). Original magnification× 20,000. Abbreviations used in figures: BM–basal membrane of endothelium; EC–endothelial cell; ER–erythrocyte; VL– vessel lumen (reprinted from [80] with permission).
Based on our many years of research efforts and experience we have found that cortical neurons from AD brain biopsies as well as animal models that mimic AD-like pathology are characterized by mitochondrial abnormalities in the cell body [2,3,46,47,77,78]. In addition, the neurons closely associated with the damaged vessels possess similar levels of ultrastructural abnormality (Figure 3). Neurons with mitochondrial lesions were seen throughout the brain cortex (Figure 4A). Our results indicate that the partially and completely damaged mitochondria are mostly located in the neuronal cell body along with lipofuscin (Figure 4A), and abnormal mitochondrial cristae were also observed close to lipofuscin granules (Figures 4B-4D). In situ hybridization of the wild type mitochondrial DNA and 8-OHG staining as a marker for the RNA oxidation in human AD brain show that wild type mtDNA (17 nm gold) is associated with severely damaged mitochondria and mitochondria-derived lysosomes, and the 8-OHG containing signals were seen throughout the neuronal cell body (Figures 5A-5C). We were not able to see any 8-OHG containing positive gold particles in the lipofuscin (Figures 5B and 5D).
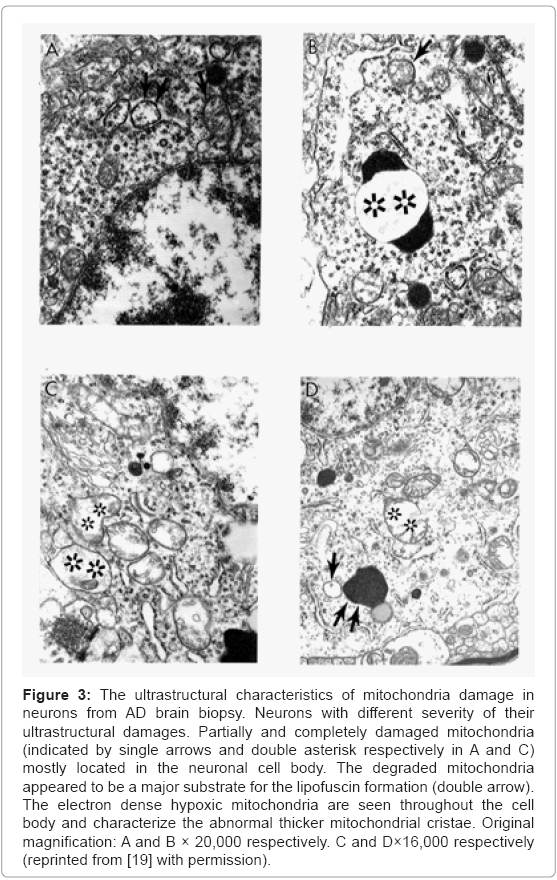
Figure 3: The ultrastructural characteristics of mitochondria damage in neurons from AD brain biopsy. Neurons with different severity of their ultrastructural damages. Partially and completely damaged mitochondria (indicated by single arrows and double asterisk respectively in A and C) mostly located in the neuronal cell body. The degraded mitochondria appeared to be a major substrate for the lipofuscin formation (double arrow). The electron dense hypoxic mitochondria are seen throughout the cell body and characterize the abnormal thicker mitochondrial cristae. Original magnification: A and B × 20,000 respectively. C and D×16,000 respectively (reprinted from [19] with permission).
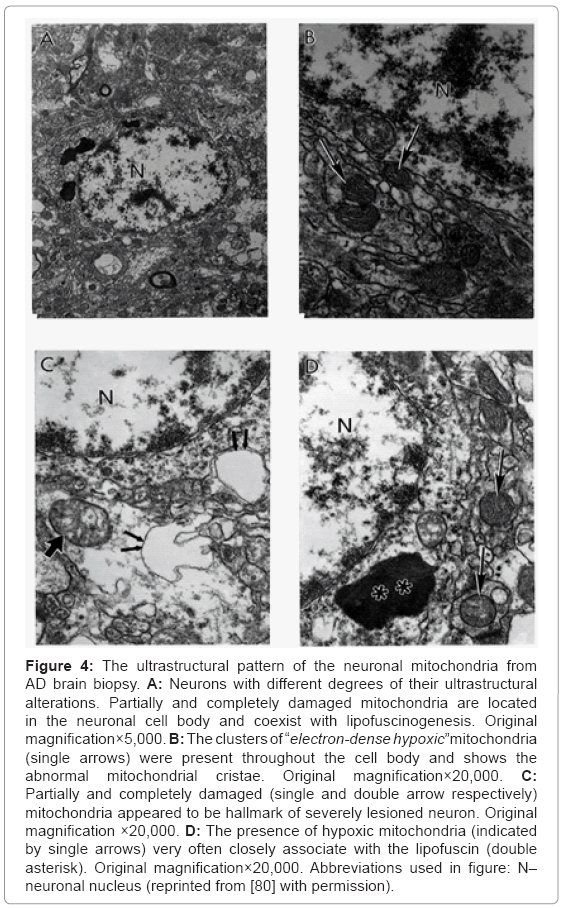
Figure 4: The ultrastructural pattern of the neuronal mitochondria from AD brain biopsy. A: Neurons with different degrees of their ultrastructural alterations. Partially and completely damaged mitochondria are located in the neuronal cell body and coexist with lipofuscinogenesis. Original magnification×5,000. B: The clusters of “electron-dense hypoxic”mitochondria (single arrows) were present throughout the cell body and shows the abnormal mitochondrial cristae. Original magnification×20,000. C: Partially and completely damaged (single and double arrow respectively) mitochondria appeared to be hallmark of severely lesioned neuron. Original magnification ×20,000. D: The presence of hypoxic mitochondria (indicated by single arrows) very often closely associate with the lipofuscin (double asterisk). Original magnification×20,000. Abbreviations used in figure: N– neuronal nucleus (reprinted from [80] with permission).
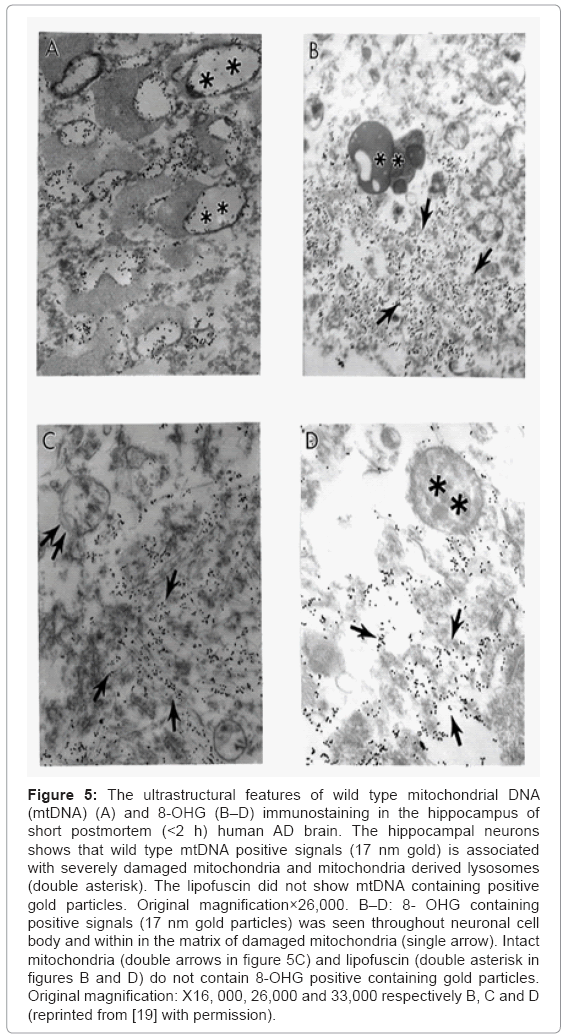
Figure 5: The ultrastructural features of wild type mitochondrial DNA (mtDNA) (A) and 8-OHG (B–D) immunostaining in the hippocampus of short postmortem (<2 h) human AD brain. The hippocampal neurons shows that wild type mtDNA positive signals (17 nm gold) is associated with severely damaged mitochondria and mitochondria derived lysosomes (double asterisk). The lipofuscin did not show mtDNA containing positive gold particles. Original magnification×26,000. B–D: 8- OHG containing positive signals (17 nm gold particles) was seen throughout neuronal cell body and within in the matrix of damaged mitochondria (single arrow). Intact mitochondria (double arrows in figure 5C) and lipofuscin (double asterisk in figures B and D) do not contain 8-OHG positive containing gold particles. Original magnification: X16, 000, 26,000 and 33,000 respectively B, C and D (reprinted from [19] with permission).
Detailed analysis of the hippocampal neurons showed that the wild type (Figures 6A and 6B), 5 kb deleted mtDNA (Figure 6C), and COX immunoreactivity (Figure 6D) were seen in the matrix of the completely damaged mitochondria and/or mitochondria derived lysosomes. The 5 kb deleted mtDNA containing gold particles (17 nm) can be seen within these mitochondria-derived lysosomes (Figure 6C). Further, these mitochondria also display COX positive immunogold particles in their matrix (Figure 6D).
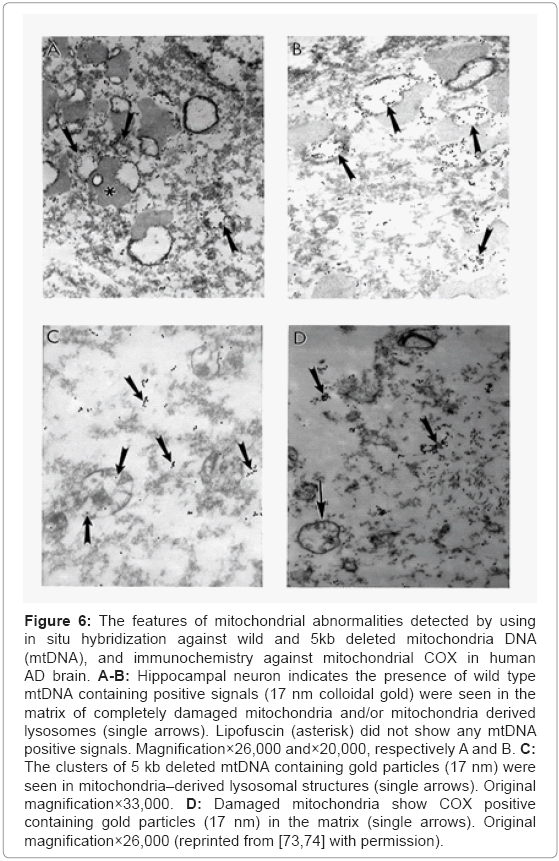
Figure 6: The features of mitochondrial abnormalities detected by using in situ hybridization against wild and 5kb deleted mitochondria DNA (mtDNA), and immunochemistry against mitochondrial COX in human AD brain. A-B: Hippocampal neuron indicates the presence of wild type mtDNA containing positive signals (17 nm colloidal gold) were seen in the matrix of completely damaged mitochondria and/or mitochondria derived lysosomes (single arrows). Lipofuscin (asterisk) did not show any mtDNA positive signals. Magnification×26,000 and×20,000, respectively A and B. C: The clusters of 5 kb deleted mtDNA containing gold particles (17 nm) were seen in mitochondria–derived lysosomal structures (single arrows). Original magnification×33,000. D: Damaged mitochondria show COX positive containing gold particles (17 nm) in the matrix (single arrows). Original magnification×26,000 (reprinted from [73,74] with permission).
Mitochondrial lesions and lipofuscinogenesis were also seen in brain tissues. The AD glial cells show accumulation of lipofuscin granules. Further, glial cells also show intracellular amyloid deposits and mitochondria derived lysosomes.
We have recently reported that aging, by itself, induces damage to mitochondria. However, the key difference between the age-matched and AD cases appear to be the extent of the mitochondrial damage [2,3,14,70,71,73].
The role for NO-dependent process is quite clear in AD pathogenesis and remodeling of cortical cholinergic system through degradation of mature nerve growth factor (NGF) in AD. It is also well established that the cortical cholinergic system plays a crucial role in cognitive processing and memory formation [79,80]. Pharmacological evidence of cholinergic atrophy and metastasis depends on matrix metalloproteinase’s in both diseases. In an activity-dependent manner NGF precursor forms proNGF, along with the convertases and proteases necessary to form mature NGF (mNGF) and to degrade the free, unbound mNGF by serine protease involved the matrix metalloproteinase 9 (MMP-9) [81]. However, the exact cellular mechanisms behind tumor vascular growth and the relationship to NO oxidation products, such as nitrotyrosine products, lipid peroxidation, as well as mitochondrial DNA (mtDNA) deletion remains unknown [9,12].
Endothelin signaling is speculated to be involved in cell differentiation, proliferation, migration, and angiogenesis in tumors [82]. It has been demonstrated that ET-1 is overexpressed in various tumor tissues [83], including prostate tumor and high grade prostatic intraepithelial neoplasia [84], breast cancer [85], lung tumor [86], and colorectal metastatic liver cancer [6,9,76]. Expression of mRNA of ET- 1, ET-RA, and ET-RB were detected in ovarian carcinoma cell lines HEY and OVCA 433 by RT-PCR, and secreted ET-1 was detected in the culture media by ELISA [83]. ET-1 and ET-RA are overexpressed in canine ovary tumors [87], which is consistent with the function of ET-RA signaling induced by ET-1 that is involved in cell proliferation [88].
Although antagonists of endothelin receptors for the treatment of cancer are not in clinical development, specific peptide-based antagonists of ET-RA and ET-RB have been used in cancer studies. Cancer cell proliferation was reported to be inhibited when ET-RA was specifically blocked in vitro and in vivo in colorectal cancer cell lines [89]. When orally active high affinity ET-RA antagonist ZD4054, which has no detectable affinity for ET-RB, was applied in vitro, it inhibited ET-1 induced proliferation of human pre-osteoblast cells [90], human ovarian carcinoma cell lines HEY and OVCA 433 [83], and further demonstrated ET-RA was involved in signaling in cancer cell proliferation.
It has been well documented that the tumor vessels characterizes the absence of perivascular nerves and suggests that endothelial derived vasoactive substances (NO and ET-1) may be the key factors in controlling tumor blood flow and tumor metastases [6,67,77]. We have demonstrated the qualitative and quantitative characteristics of the ultrastructural distribution of different NOS isoforms and ET-1 immunoreactivity in human colorectal metastatic tumor liver using preembedding peroxidase-anti-peroxidase (PAP) and post-embedding immunoelectron microscopic triple gold labeling techniques [6,67,76].
Electron Microscopic PAP techniques investigation of the distribution of NOS1 immunolabeling in control (Figure 7A) and metastatic colorectal cancer liver tumor tissues (Figures 7B-7D) showed that NOS1 immunopositive EC were seen in control liver microvessels. The tumor vessel endothelium showed no positive staining against for NOS1 antibody (Figures 7A and 7B). However, very often the presence of NOS1 immunopositive white blood cells that adhered to vessel endothelium in tumor growth regions was observed (Figure 7C). Moreover, NOS1 immunopositive staining myofibroblast (smooth muscle cell) was also seen in metastatic liver tumor tissues (Figure 7D).
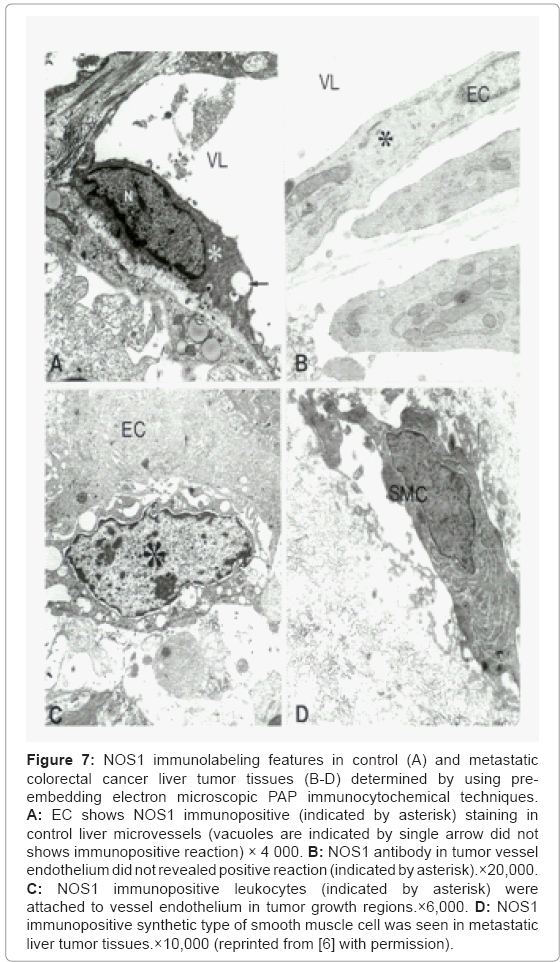
Figure 7: NOS1 immunolabeling features in control (A) and metastatic colorectal cancer liver tumor tissues (B-D) determined by using preembedding electron microscopic PAP immunocytochemical techniques. A: EC shows NOS1 immunopositive (indicated by asterisk) staining in control liver microvessels (vacuoles are indicated by single arrow did not shows immunopositive reaction) × 4 000. B: NOS1 antibody in tumor vessel endothelium did not revealed positive reaction (indicated by asterisk).×20,000. C: NOS1 immunopositive leukocytes (indicated by asterisk) were attached to vessel endothelium in tumor growth regions.×6,000. D: NOS1 immunopositive synthetic type of smooth muscle cell was seen in metastatic liver tumor tissues.×10,000 (reprinted from [6] with permission).
The electron microscopy PAP immunocytochemical labeling of inducible NOS (NOS2) immunoreactivity in metastatic liver tumor tissues showed that tumor vessel EC was positively stained with NOS2 (Figure 8A). A high intensity of NOS2 immunopositive precipitate accumulation close to the luminal plasmalemma of the vascular EC in the tumor growth region were also seen figure 8B, indicating the elevated tissue levels of NO and perhaps ET-1 as well [6,9,76]. The presence of NOS2 immunopositive hepatocytes and myofibroblastlike cells throughout the tumor growth area appeared to be permanent markers of the tumor growth and metastases (Figure 8C). However, the lipid-contained areas of the cellular matrix were free from PAP immunopositive containing precipitate visualized by using preembedding immunocytochemical techniques (Figures 8C and 8D).
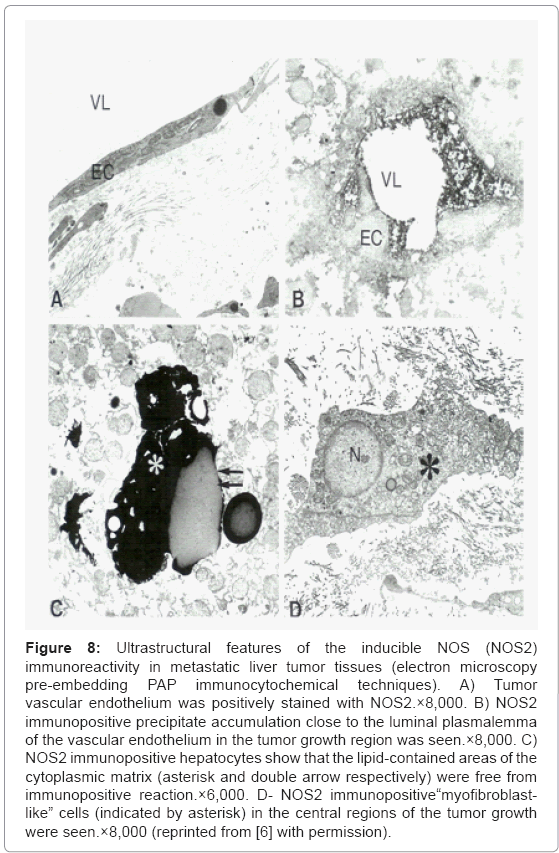
Figure 8: Ultrastructural features of the inducible NOS (NOS2) immunoreactivity in metastatic liver tumor tissues (electron microscopy pre-embedding PAP immunocytochemical techniques). A) Tumor vascular endothelium was positively stained with NOS2.×8,000. B) NOS2 immunopositive precipitate accumulation close to the luminal plasmalemma of the vascular endothelium in the tumor growth region was seen.×8,000. C) NOS2 immunopositive hepatocytes show that the lipid-contained areas of the cytoplasmic matrix (asterisk and double arrow respectively) were free from immunopositive reaction.×6,000. D- NOS2 immunopositive“myofibroblastlike” cells (indicated by asterisk) in the central regions of the tumor growth were seen.×8,000 (reprinted from [6] with permission).
Ultrastructural features of endothelial specific NOS (eNOS or NOS3) labeling in control (Figure 9A) and metastatic liver tumors tissues (Figures 9B-9D) determined by using PAP method shows the presence a large number of NOS3 immunopositive EC were seen only in control liver microvessels. NOS3 immunostaining was absent in EC in tumor vessels (Figures 9B and 9C). However, the presence of NOS3 immunopositive hepatocytes in metastatic liver tumors were seen throughout tumor growth regions. Lipid-enriched areas were free from NOS3 immunopositive precipitate (Figure 9D).
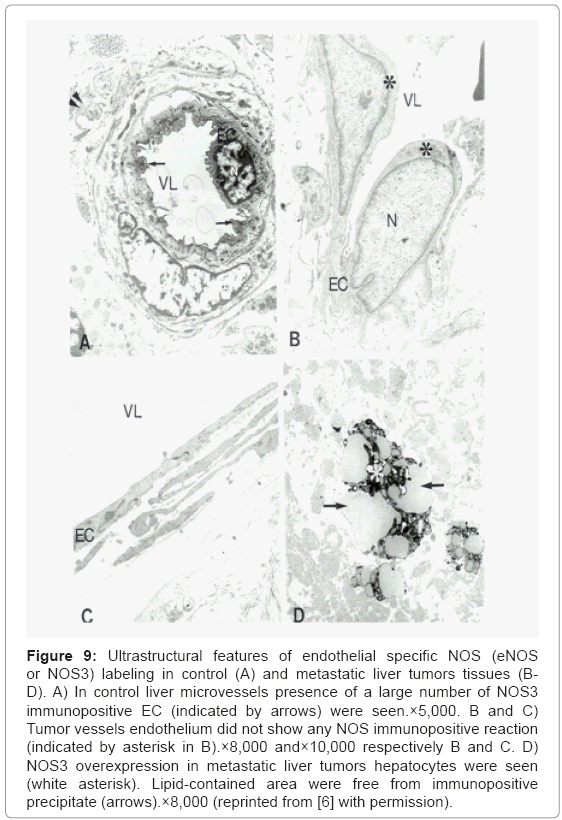
Figure 9: Ultrastructural features of endothelial specific NOS (eNOS or NOS3) labeling in control (A) and metastatic liver tumors tissues (BD). A) In control liver microvessels presence of a large number of NOS3 immunopositive EC (indicated by arrows) were seen.×5,000. B and C) Tumor vessels endothelium did not show any NOS immunopositive reaction (indicated by asterisk in B).×8,000 and×10,000 respectively B and C. D) NOS3 overexpression in metastatic liver tumors hepatocytes were seen (white asterisk). Lipid-contained area were free from immunopositive precipitate (arrows).×8,000 (reprinted from [6] with permission).
Implications of post-embedding triple immunogold labeling techniques showed that the clusters of NOS2 positive, but not NOS3 and ET-1 immunopositive containing gold particles were seen in tumor vessel endothelium (Figure 10A). The presence of a NOS1 positive containing gold particles were seen in the matrix of lipid laden hepatocytes in tumor growth area, and coexists with the presence of a NOS3 and ET-1 positive gold particles (Figure 10B). Very often the clusters of ET-1 but not constitutive isoforms of NOS (NOS1 and NOS3) positive gold particles in the cell cytoplasmic matrix, especially in the matrix of mitochondria of hepatocytes were seen in figure 10C. The metastatic liver microvessels endothelium prepared as negative controls (through omission of the primary antibody) very occasionally showed only the presence of single gold particles (Figure 10D). This observation highlights mitochondria as a critical constituent responsible for cell viability, which can be considered as a new research focus and of new diagnostic criteria for the earlier detection of tumors as well as treatment strategies at least in some tumors. However, further studies need to be carried out in order to clarify the exact nature of these relationships during the metastases and growth of primary and/ or metastatic cancers [6,67,76].
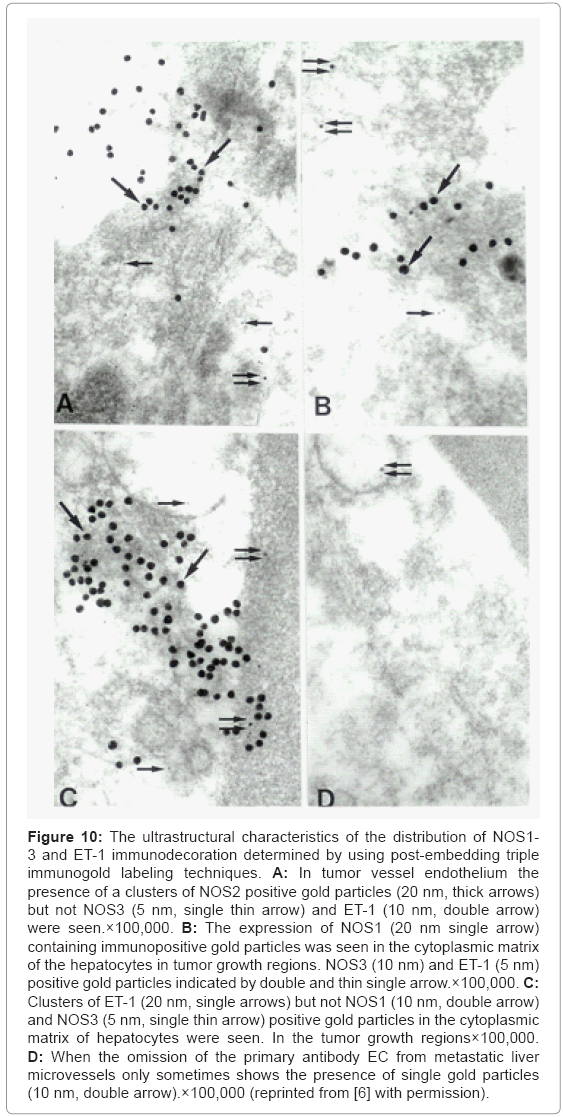
Figure 10: The ultrastructural characteristics of the distribution of NOS1- 3 and ET-1 immunodecoration determined by using post-embedding triple immunogold labeling techniques. A: In tumor vessel endothelium the presence of a clusters of NOS2 positive gold particles (20 nm, thick arrows) but not NOS3 (5 nm, single thin arrow) and ET-1 (10 nm, double arrow) were seen.×100,000. B: The expression of NOS1 (20 nm single arrow) containing immunopositive gold particles was seen in the cytoplasmic matrix of the hepatocytes in tumor growth regions. NOS3 (10 nm) and ET-1 (5 nm) positive gold particles indicated by double and thin single arrow.×100,000. C: Clusters of ET-1 (20 nm, single arrows) but not NOS1 (10 nm, double arrow) and NOS3 (5 nm, single thin arrow) positive gold particles in the cytoplasmic matrix of hepatocytes were seen. In the tumor growth regions×100,000. D: When the omission of the primary antibody EC from metastatic liver microvessels only sometimes shows the presence of single gold particles (10 nm, double arrow).×100,000 (reprinted from [6] with permission).
Under the influence of continuing stimuli, or mitotic drive which are capable to upregulate cyclin-dependent kinases (Cdks) and their cognate activating cyclins to orchestrate DNA replication, cytoskeletal reorganization and cellular metabolism that outcomes as a cell proliferation. Hormonal signals from luteinizing hormone and other hormones can contribute to this mitotic drive and pathology such as AD [91]. Mitotic drive and the orderly progression through cell cycle, involve cyclins and Cdks which form complexes that are able to phosphoregulate a wide variety of substrates [92]. However, some of researcher reported that extrinsic mitotic pressures, and proper cell cycle progression, can also involve desensitization. In addition, the recruitment of cell cycle elements creating intrinsic mitotic pressures on developmentally fated cells can lead to pathogenic states such as cancer and AD. Recent reports showed direct associations with AD and ADrelated cytoskeletal pathology [93], that most likely may be involved in aberrant neuronal sprouting response [94] or other activity [95-97]. In other hand, AD, inflammation, and cancer are intimately associated. In the same regard, mitotic drive can be derived from the inflammatory signals, oxidative stress and/or related excitatory stressors [98-100]. Strong literature evidence support exists in literature for an AD-cell cycle-associated emergence from a quiescent state and researchers have looked at it as a recapitulation or vestige of an evolutionarily conserved process [101,102]. In addition, recent study reported possibility reversion in AD pathology by using anticancer drugs [103]. Moreover, AD-associated proteins and the cell cycle activation from mitotic drive are intimately linked to both tau proteins. Because tau is the major protein component of NFT an intracellular hallmark of AD, as well as to Aβ, the extracellular lesion associated with the disease [104-107], and the increased phosphorylation of the microtubule associated tau protein that destabilizes microtubular dynamics and leads to neuronal dysfunction and therefore memory decline [108,109]. Perhaps the strongest case for the oncogenic relation between cancer and AD are best elucidated in our recent report [69]. Most likely this phenomenon associated with oncogenic transduction pathway activation has attracted the interest of researcher for a few years now. Another strategy for tumor treatment has focused on the inhibition of tumor angiogenesis. It has been well established that angiogenesis is a critical event in tumor growth and metastasis [110]. Increased NO production may selectively support mutant p53 cells and may also contribute to tumor angiogenesis by upregulation of vascular endothelial growth factors. We hypothesize that mitochondrial involvement in this cascade is a major factor in the control and regulation of tumor growth and metastasis. Therefore, it is very important to understand the oncogenic factors in a context geared towards medical applications in order to mediate the chronic effects of cancer [111]. Although polyphenolic antioxidants may be beneficial in the prevention of certain diseases including cancer and neurodegenerative diseases, there are questions regarding their potential harmful effects due to their involvement in glycation, and the advanced glycation products (AGEs) from these glycation reactions may exert cellular toxicity. Therefore it is too early to unambiguously prove the beneficial effects of polyphenolic antioxidants on the age-related diseases. We have recently critically examined general antioxidant compounds in health, disease and aging with hope that a better understanding of the many mechanisms involved with these diverse compounds may lead to better health and novel treatment approaches for age-related diseases including the cancer and neurodegeneration [112].
Research data and literature evidence presented in this paper indicates that we were able to demonstrate the ultrastructural features of the mitochondrial pathology by using classical electron microscopy and in situ hybridization at the ultrastructural levels in human AD, AD-like pathology in the animals that mimics human AD, human colorectal cancer metastases into the liver and malignant and benign brain tumors. We have found that the abnormal mitochondria (e.g. mitochondria with electron dense matrix, mitochondrial-derived lysosomes) and their residues (lipofuscin) appear to be features of hippocampal neurons in human AD, aged Tg(+) mice, 2 vessel occlusion model of the brain hypoperfusion, and malignant primary and metastatic cancer.
In situ hybridization analysis of mouse and human mtDNA probes show a large number of deleted mtDNA in human AD and in animal models that mimic human AD. In addition, vessel endothelium from AD brain and tumor tissues shows damage of mitochondrion cristae and positive immunostaining against inducible NOS and ET-1. The lipid laden tumor cells and surrounding cells often show a different degree of mitochondria abnormality. Moreover, biopsies from AD brains, and perfused brain samples from the animal models that mimics human AD as well as tissues from the cancer cases were dominated by abnormal mitochondria, i.e., 3 to 4 fold higher in AD as compared to controls. Only hippocampal, vulnerable cortical neurons and malignant cancer tissues showed immunopositive staining for RNA oxidation markers visualized by using 8-OHG-staining, NOSs, and oxidative stress markers. The mitochondrial DNA overproliferation and deletion detected by using cytological techniques suggests that dysregulation of the cell cycle is a hallmark of neoplasm. Our studies show that oxidative stress markers are abundant in AD brain and malignant cancer cells indicating that chronic oxidative stress induced mitochondrial DNA overproliferation and/or deletion plays a primary pathogenic role in the pathogenesis of AD and cancer. Therefore, common features of mitochondrial abnormality were seen on the brain during tumorigenesis and AD, indicating that mitochondrial DNA overproliferation and/or deletion are the key initiating factors for development, maturation, and progression of neurodegeneration as well as tumor growth and/or metastases. In addition, our previous experimental and ongoing clinical studies showed the preservation and improvement of cognitive tasks in animal models that mimics human AD-like pathology as well as depressed and demented patients after 24, 36 and 60 month follow up of combined pharmacological (the combination of the diseases and mitochondrion specific compounds) and non-pharmacological treatment [5,16]. The treatments included using antidepressants, cholinesterase inhibitors, NMDA antagonists, multivitamins, and antioxidants including alpha-lipoic acid and N-acetyl-L-carnitine. After 60 months of treatment, above base line performance of all the tasks was the same as or above the baseline. The MMSE, Cognistat–Attention, Cognistat–Judgment, and RFFT-Total Unique Designs demonstrated significant improvement in memory [5,16]. Our results also demonstrate the arrest in cognitive decline in demented patients for 60 months [5,16]. Therefore, the continues chronic cellular hypoperfusion and oxidative stress in brain cellular compartments can stimulate the expression of extremely labile active molecules such as NO and NOSs enzymes. The idea behind this is that brain microvessels in the endothelium increases the accumulation of oxidative stress induced products or their derivatives [1-3,5,16,23,36-38,58,59,54,69-74,76,78,113,104,111-114]. Consequently, NO and ROS contribute to BBB dysfunction and brain parenchymal cell damage [4]. Tracking the molecular mechanisms of these processes may provide clues for the novel therapeutic targets for cerebrovascular diseases such as stroke and their consequence which are manifested as a mental retardation and memory decline that appear to be a main clinical sign of AD patients [1-3,5,7,9,11,13].
We speculate that mitochondrial involvement may play a significant role in the etiopathogenesis of cancer. Further, considerable evidence also directs towards an oncogene parallel between AD and cancer. Studies also demonstrate a potential relationship between anticancer drugs and early explorations of mode of action, i.e. inhibition of AD-like pathology in transgenic mice. Our research finding indicates that severity of the cancer growth, metastasis as well as brain pathology seen in AD and/or animals that mimic human AD correlate with the degree of mitochondrial abnormalities. Exploration of the role of NO dependent mitochondrial DNA overproliferation/deletions as well as mitochondrial enzyme activity during tumor growth/metastases may provide important insights that affect aging and biochemical perturbation on mitochondrial DNA integrity [2-3,6,67]. However, still one of the big challenges for treatment of neurodegenerative diseases and cancer appeared to be delivering drugs into the injury affected tissues. Our future goals are aimed to determine that injection of silver nanoparticles in the brain lead to leaking on the inter-endothelial contact and luminal plasma membrane, and therefore elucidate the possibility of penetrating into the cerebrovascular, neuronal, and glial cell which are especially damaged in AD and/or cancer. We are confident that our research involving the conjugation of the silver nanoparticles with mitochondrion specific and or their derivative drugs would help not only to diminish the lesions that occur in AD and/or tumor tissues, but also allow for future applications of a combined, integrative treatment model in clinical practices [2,5,7,9,13,15-16,21].
This study was partially supported by “GALLY” International Biomedical Research Consulting LLC, San Antonio, TX, USA (for G. Aliev), NIH (for Hector H. Palacios) and EuroEspes Biomedical Research Center, Institute for CNS Disorders and Genomic Medicine & Camilo José Cela University, La Coruña, Spain (for P. Cacabelos and R. Cacabelos).