Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Rapid Communication - (2016) Volume 9, Issue 2
Motivation: MLVA (Multiple Loci VNTR Analysis) is a typing method used today to characterize several major pathogens such as Brucella, Mycobacterium tuberculosis or Salmonella. It takes advantage from the comparison of the size of specific genomic loci constituted by tandem repeat sequences. Unfortunately the raw size estimate is instrument dependent and consequently the results obtained cannot be compared without normalization between laboratories involved in disease monitoring, surveillance and official controls.
Results: To overcome this problem we developed a workflow tool, MLVA_normalizer, conceived to normalize MLVA results. This normalization workflow tool is designed to be applied to any bacterial genera and does not depend on the MLVA protocol used.
Availability: MLVA_normalizer is available under the GNU general public license (version 2, June 1991). Source code is available at: https://github.com/afelten-Anses/MLVA_normalizer.
Keywords: MLVA, Workflow, Python 2.7 langage, Normalisation, Laboratory data exchange
Multiple Loci VNTR Analysis (MLVA) is a method employed for typing microorganisms, such as pathogenic bacteria. This analysis takes advantage of the genomic polymorphism of tandemly repeated DNA sequences called VNTRs (variable-number tandem repeats) [1]. Use of this typing method has grown significantly over the past decade in response to numerous limitations encountered by the standard typing methods and thanks to its ease and speed of application in the field. For Salmonella, for example, the standard typing method is pulsed-field gel electrophoresis (PFGE). This method requires specific technical expertise, is labor-intensive and provides insufficient discrimination within several serovars [2]. As a result, on account of its capacity to differentiate closely related strains, MLVA has become a major first line typing tool for a number of pathogens such as Mycobacterium tuberculosis [3], Bacillus anthracis [4], Brucella [5], Staphylococcus aureus [6], Salmonella [7] and Shiga toxin-producing Escherichia coli [8].
In a typical MLVA assay, a number of VNTR loci are amplified by polymerase chain reaction (PCR) so that the size of each locus can be measured, usually by electrophoresis of the amplification products together with reference DNA fragments (known as DNA size markers). From this size, the number of tandem repeat units (TRs) at each locus can be deduced. The number of TRs for each locus is collected in a code that becomes the MLVA profile of the organism analyzed.
Unfortunately, depending on the electrophoresis equipment used, errors can appear in the estimation of the size of VNTRs. The raw size estimate, for the same DNA fragment, is indeed instrument dependent and can results in differences in the attribution of allele numbers. Consequently the MLVA profile obtained for the same strain can differ between laboratories. The capillary equipment usually come with software enabling the allele calling from size bins but their setting is often elaborated and limited in term of correcting the raw data. It’s why paying softwares exist and ECDC published in 2011 for Salmonella enterica subsp. enterica serovar typhimurium, a pathogen of major interest in public health, an Excel file in order to standardize data from laboratories participating at interlaboratory comparison study for the MLVA analysis of this pathogen. In the context of major health problems or foodborne pathogens, precise and rapid characterization is fundamental for the implementation, strengthening and evaluation of health and sanitary policies. We built MLVA_normalizer workflow to ensure accurate MLVA results to aid data exchange between laboratories involved in disease monitoring and surveillance.
The MLVA_normalizer workflow was built using Python 2.7. The algorithm was inspired by an Excel file published in ECDC’s Laboratory Standard Operating Procedure for MLVA of Salmonella enterica serotype typhimurium (2011). The essential prerequisite for running MLVA_normalizer workflow, as in any assay and quality control, is to have a reference strain panel for which the lengths of VNTR loci have been confirmed by sequencing. For several pathogens, such as Mycobacterium tuberculosis [3], Salmonella serovar typhimurium [9] and enteritidis [10], this reference strain table already exists. It includes the name of the reference strains, the correct MLVA profile and the real length of each VNTR locus analyzed. Any laboratory wishing to use MLVA_normalizer workflow must possess the appropriate panel of reference strains and analyze them together with the sample strains. The electrophoresis data (measured length) must be collected (both for the reference strain and for the sample strain) and organized in two different input files: i/ reference.txt input file, containing the data for the reference strain (Figure 1) and ii/ capillary_electrophoresis.txt input file, with data for the sample strain. The capillary_electrophoresis.txt input file must be compiled by the user as in Figure 1 and the name of the VNTR must be typographically the same as that of the reference.txt input file. The two input files must be in (.txt) format to run in MLVA_normalizer workflow. The algorithm follows three steps (Figure 1):

Figure 1: Input and output data files and steps of the MLVA normalization workflow algorithm. a: Name of the VNTR locus; b: Length of the regions upstream and downstream of the VNTR locus; c: Length of the tandem repeat unit; d: Length measured by electrophoresis including instrument error; e: Real length confirmed by sequencing.
i) Creation of a correction matrix: This matrix is built with the data present in the reference.txt input file. The difference between the real corrected length and the measured length is calculated for each VNTR for each reference strain. After that, a sliding average of the differences is calculated to obtain the best correction fit for each VNTR as 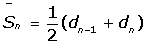 [11]. The best correction fit
[11]. The best correction fit 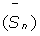 corresponds to the average between the difference calculated for one repeat unit and the previous one (dn-1 and dn).
corresponds to the average between the difference calculated for one repeat unit and the previous one (dn-1 and dn).
ii) Creation of a normalization matrix: This matrix is built with the correction matrix obtained above and the capillary_electrophoresis.txt input file. The measured lengths, obtained for the sample strains, are corrected with the correction fit to obtain normalized results.
iii) Choosing the correct number of tandem repeats: This step is performed by comparing, for each VNTR, the normalized results obtained above with the corrected length recorded in the reference.txt input file. Four possibilities are considered: i) if the normalized result gives the same length as the flanking size (Figure 1), no tandem repeats are present and the MLVA profile will be 0. ii) If the normalized result is equal to 0, no PCR product is present, so the MLVA profile will be -2 in accordance with a previously published convention [1]. iii) If the normalized result is within two base pairs (plus or minus) of the corrected length, then the same number of tandem repeats of the reference strain will be assigned (see “strain_1 in Results.txt”, Figure 1: (110-2) <111< (110+2) =>1 TR as in the reference strain). iv) If the condition in point iii/ is not met, a warning (“Check”) is displayed, meaning that the value obtained must be verified. Three checking messages can appear they are illustrated in the results_new_algo.txt file. For example, for the VNTR called STTR9, the checking message [-∞; 2.0] means that the measured fragment is smaller than what was previously found, in this case: 2 repeated units. In the same way, the message [9.0;+∞] means that the fragment size is longer compared to what was previously found; in this case, the fragment size exceeds the size of 9 united repeats, the longer size observed. Finally the message [3.0; 4.0] indicates that the fragment size cannot be assigned to an allele, in this case the alleles 3 and 4. This could depend on a lot of factors and the analysis should be done again.
The results.txt output file will contain the normalized MLVA profiles for the strains analyzed. The Python scripts, user and technical documentations can be found on gitub (https://github.com/afelten-Anses/MLVA_normalizer).
To validate the workflow we compared 215 MLVA profiles for Salmonella enterica serovar typhimurium with results from the ECDC spreadsheet cited above. Up today, in our laboratory we have analyzed more than 800 MLVA profiles, for Salmonella typhimurium, enteritidis and dublin with this workflow.
The use of this workflow is laid to the availability of a panel of strains (known as reference strains) for which the size of alleles in every locus is known. The importance to have this set of calibration strains is largely explained by Larson in 2013 [12]. This author used 20 international laboratories analyses to proof that without a set of reference strains it is not possible to obtain comparable results between laboratories [12]. The workflow proposed in this study can name alleles in new isolates only if the size of these alleles is in the reference set. If the size of the allele is beyond the set of alleles of reference strains, this allele is considered as “new” and must be verified by sequencing. Another condition to use MLVA_normalizer is lied to the distance between alleles: it must be higher than five base pairs (pb). This is due to the fact that two bp (plus or minus) are the lapse of error accepted in calculation to can name alleles. Hence the workflow fails if the typing scheme uses repeats shorter than five bp. It is the case for some VNTR as SAL20 for Salmonella and O157-3, O157-25 or O157-17 for E.coli [8,13]. However, an international scientific consensus exists exhorting to not include in a subtyping protocol, repeat units shorter than five bp because of the limitations in sizing reproducibility in capillary electrophoresis platforms [1,8].
MLVA_normalizer is easy-to-use tool that computes reference values and electrophoresis data stored in .txt format in a manner more prone to automatization (high throughput, large data sets) and versatility (no manual filling of spreadsheets required) compared to existing tools such as the ECDC spreadsheet. This tool can evolve with the user. Indeed, even if we propose .txt files for the analysis of Salmonella, MLVA_normalizer can be used for whatever other organism at condition to have a panel of reference strains (as discussed upper in the document) and to analyze repeated units which size is higher than five bp.
MLVA_normalizer is finally a free tool that can allows, the normalization of MLVA results whatever the organism analyzed and protocol used. In the Food Safety Laboratory of ANSES, we use the MLVA_normalizer workflow routinely to normalize the MLVA profiles of Salmonella strains such as S. typhimurium, Enteritidis and Dublin for monitoring and surveillance, investigations of outbreaks and official controls.
Research reported in this publication was supported by funds from the Ministère de l’Agriculture, de l’Agroalimentaire et de la Forêt and the Association de Coordination Technique pour l’Industrie Agro-Alimentaire (ACTIA-UMT ARMADA).
None declared.