Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Research Article - (2016) Volume 5, Issue 3
Saccharomyces cerevisiae is the most widely used yeast in research and industries, however the downstream processes for its protein production are costly. This study attempted to find out a simple way to predict the success rate of protein purification with amino acid features. Logistic regression and neural network model were used to test each of 535 amino acid features one by one against the purification state of 1294 expressed proteins from S. cerevisiae, of which 870 were purified. The results show that the predictive performance of neural network is more powerful than that of logistic regression. Some amino acid features are useful to predict the purification tendency of proteins, and the varying amino acid features perform better as demonstrated by very high sensitivity accompanied with low specificity. Moreover, the S. cerevisiae proteins with a high predictable portion of amino acid pairs have higher accuracy of purification prediction than those with a low predictable portion. Thus, the success rate of purification of S. cerevisiae proteins can be predicted using neural network based on protein sequence information. This simple prediction process can provide a concept about the probability of a protein is purified, which should be helpful to overcome blindfold experiments and enhance the production of designed proteins.
Keywords: Amino acid feature, Protein purification, Prediction, S. cerevisiae
Saccharomyces cerevisiae is the most useful yeast for humans, and since ancient times it has been widely used in winemaking, baking, and brewing. Over the past two decades, efforts have been made to reduce alcohol levels in wines through rational and evolutionary engineering of S. cerevisiae in order to maintain consumer health, prevention policies, the effectiveness of the fermentation and wine sensorial quality [1]. On the other hand, genetic engineering has provided non-conventional yeast species with unusual tolerance in order to produce high yields of liquid fuels and commodity chemicals from lignocellulosic biomass [2,3]. As a unicellular eukaryotic model, S. cerevisiae is one of the most intensively studied organisms in molecular and cell biology [4], and generates major breakthroughs in understanding of the mechanisms of cellular and molecular processes [5-7]. Although it has been used in fundamental and applied researches for long times, the interests in S. cerevisiae do not decrease but increase recently. For example, S. cerevisiae is used as a model to study Alzheimer’s Disease [8], Parkinson’s disease [9] and mitochondrial diseases [10].
Cell-free protein synthesis based on S. cerevisiae is a versatile technique to produce proteins on-demand [11]. As an important industrial workhorse in biotechnology, the production of proteins from S. cerevisiae has been accelerated by the upcoming demand for sustainable processes and renewable raw materials [12]. Some suitable expression systems were identified and optimized, and enhance the production of recombinant proteins for medical or industrial uses [13]. Consequently, it is important to facilitate purification process to obtain the recombinant proteins at desirable purity, whereas there are still significant challenges because many steps can influence purification, such as affinity chromatography, precipitation, protecting of recombinant proteins from degradation with stabilizer, centrifugation, etc.
Nowadays, we are in the era of big-data and the question raised here is how to use available data to improve protein purification. This could possibly be resolved because many expressed and purified proteins are available from databank, so the success rate of purification can be studied through modeling. Because proteins are composed of amino acids, amino acid features should have certain relationships with protein purification. For many expressed proteins, their amino acid composition, primary structure, and sometimes 3-dimensional structure are clearly documented, which provide useful information to analyze the success rate of purification of proteins of interest. The predicted result from modeling will give researchers a concept on what a chance a protein of interest can be successfully purified before conducting the experiment. Such modeling analyses have been done in predicting crystallization propensity of different proteins [14-19] but yet been applied to purification of proteins. Thus, this study aimed at analyzing the purification success of S. cerevisiae protein by means of modeling.
Model output
The success rate of protein purification of S. cerevisiae is analyzed using the number of purified proteins versus that of expressed proteins. Before 2012, 870 proteins from S. cerevisiae were successfully purified among 1294 proteins that were successfully expressed [20]. The success rate of purified proteins in this regard constructs so-called yes-no event, i.e., 1 and 0 event mathematically. The purified proteins were classified as 1 while the expressed ones were classified as 0, which severed as model output for the prediction and their details were listed in Table S1 of Supplementary Information.
Model inputs
Constant features: Currently, 544 amino acid features are documented in AA-Index [21], and each feature contains 20 constant values for 20 types of amino acids. For example, a physicochemical feature of amino acid (FAUJ880108) describes the localized electrical effect [22]. Actually, not every feature has 20 values, so 531 features were used as model inputs one-by-one, including 40 composition features, 218 physicochemical features, and 273 features related to second structure (Table S2 of Supplementary Information). An important point is that these 531 amino acid features are constant values regardless amino acid’s position, neighbor, etc. Table 1 showed how the model inputs were different between two S. cerevisiae proteins (accession numbers in UniProtKB P38765 and Q12380) as an example. They have the same length of 294 amino acids but their amino acid compositions are different (columns 2 and 3 in Table 1). When using the amino acid feature FAUJ880108 as inputs, its values were constant (column 4) regardless the difference between two proteins. In order to overcome this limit, these values could be weighted by the composition of amino acids (columns 5 and 6), by which the constant features are subject to the amino acid compositions.
| Amino acid | No. | FAUJ880108 | FAUJ880108 ´ No. | CC, % | FC, % | DP | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P1 | P2 | P1 | P2 | P1 | P2 | P1 | P2 | P1 | P2 | |
| A | 28 | 8 | -0.01 | -0.01 | -0.28 | -0.08 | 9.52 | 2.72 | 7.65 | 5.24 | 0.0115 | 0.17 |
| R | 6 | 12 | 0.04 | 0.04 | 0.24 | 0.48 | 2.04 | 4.08 | 6.03 | 7.64 | 0.0386 | 0.02 |
| N | 15 | 15 | 0.06 | 0.06 | 0.9 | 0.9 | 5.1 | 5.1 | 4.62 | 4.45 | 0.0374 | 0.16 |
| D | 12 | 15 | 0.15 | 0.15 | 1.8 | 2.25 | 4.08 | 5.1 | 4.57 | 3.36 | 0.0399 | 0.16 |
| C | 4 | 3 | 0.12 | 0.12 | 0.48 | 0.36 | 1.36 | 1.02 | 2.09 | 2.82 | 0.5625 | 0.22 |
| E | 24 | 13 | 0.05 | 0.05 | 1.2 | 0.65 | 8.16 | 4.42 | 4.16 | 3.48 | 0.0259 | 0.02 |
| Q | 7 | 9 | 0.07 | 0.07 | 0.49 | 0.63 | 2.38 | 3.06 | 3.1 | 2.75 | 0.2142 | 0.2 |
| G | 16 | 16 | 0 | 0 | 0 | 0 | 5.44 | 5.44 | 5.58 | 5.08 | 0.0568 | 0.06 |
| H | 5 | 5 | 0.08 | 0.08 | 0.4 | 0.4 | 1.7 | 1.7 | 2.76 | 3.09 | 0.384 | 0.38 |
| I | 24 | 30 | -0.01 | -0.01 | -0.24 | -0.3 | 8.16 | 10.2 | 6.23 | 6.91 | 0.0623 | 0.04 |
| L | 21 | 28 | -0.01 | -0.01 | -0.21 | -0.28 | 7.14 | 9.52 | 8.53 | 10.5 | 0.0707 | 0.01 |
| K | 24 | 19 | 0 | 0 | 0 | 0 | 8.16 | 6.46 | 4.29 | 3.84 | 0.0714 | 0.04 |
| M | 8 | 8 | 0.04 | 0.04 | 0.32 | 0.32 | 2.72 | 2.72 | 2.03 | 2.25 | 0.2243 | 0.07 |
| F | 16 | 18 | 0.03 | 0.03 | 0.48 | 0.54 | 5.44 | 6.12 | 3.09 | 3.57 | 0.0795 | 0.01 |
| P | 16 | 17 | 0 | 0 | 0 | 0 | 5.44 | 5.78 | 5.23 | 5.11 | 0.0715 | 0.07 |
| S | 17 | 28 | 0.11 | 0.11 | 1.87 | 3.08 | 5.78 | 9.52 | 7.68 | 8.28 | 0.0549 | 0.01 |
| T | 21 | 17 | 0.04 | 0.04 | 0.84 | 0.68 | 7.14 | 5.78 | 7.45 | 6.8 | 0.0371 | 0.02 |
| W | 2 | 8 | 0 | 0 | 0 | 0 | 0.68 | 2.72 | 0.62 | 0.77 | 0.5 | 0.25 |
| Y | 10 | 7 | 0.03 | 0.03 | 0.3 | 0.21 | 3.4 | 2.38 | 2.56 | 2.73 | 0.0714 | 0.02 |
| V | 18 | 18 | 0.01 | 0.01 | 0.18 | 0.18 | 6.12 | 6.12 | 7.67 | 7.18 | 0.0748 | 0.12 |
FAUJ880108 is a physicochemical feature of amino acids that describes the localized electrical effect [22]. P1 and P2 are two proteins with accession number YSG-YHR029c and YSG-YPL149w. No., Number of amino acids; CC, %, the current composition of amino acids calculated by the number of a type of amino acids divided by the total number of amino acids in a protein; FC, %, the future composition of amino acids calculated according to the mutating probability (https://www.nerc-nfb.ac.cn/calculation/fc.htm); DP, the distribution probability of amino acids can be calculated at https://www.nerc-nfb.ac.cn/calculation/dp.htm.
Table 1: Comparison of constant and dynamic amino acid features in two proteins.
Varying features: Four varying features of amino acids were used in this study and listed in the last 4 rows of Table S2. (1) The number of amino acids, which is a basic and simple varying feature for a protein as shown at columns 2 and 3 in Table 1. (2) The current composition of amino acids, which is the percentage of certain type of amino acids divided by total number of amino acids in a protein as shown at columns 2 and 3 in Table 1. (3) The future composition of amino acids, which was computed according to the mutating probability listed in Table S3 of Supplementary Information [23-26] with web computation https:// www.nerc-nfb.ac.cn/calculation/fc.htm and the two examples were showed at columns 7 and 8 in Table 1. (4) The distribution probability of amino acids, which was computed according to the equation:
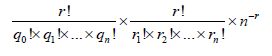
where ! is the factorial, r is the number of a type of amino acid, q is the number of partitions with the same number of amino acids and n is the number of partitions in the protein for a type of amino acid [27] with web computation https://www.nerc-nfb.ac.cn/calculation/dp.htm.
The last varying feature is the amino acid pair predictability [25,26]. Because an amino acid for constructing an amino acid pair is independent of other amino acids, probabilistic principle of multiplication can be applied to computing the predictability of amino acid pairs in a protein. Let the protein P38765 as an example, it has 294 amino acids, among them there are 28 alanines (A) and 21 threonines (T). According to the permutation, the amino acid pair AT would appear twice in this protein, 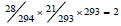 .In realty this protein does have two ATs, so the amino acid pair AT is predictable. Again according to the permutation, the amino acid pair TT would appear once,
.In realty this protein does have two ATs, so the amino acid pair AT is predictable. Again according to the permutation, the amino acid pair TT would appear once, 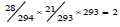 .However it appears 4 times in this protein, so the amino acid pair TT is unpredictable. By this way, all amino acid pairs can be classified as predictable or unpredictable, and their sums constitute the predictable and unpredictable portions of a protein. For this protein, its predictable and unpredictable portions are 55.25% and 44.75%. This feature is generally different from protein to protein, which can be computed at the web https://www.nerc-nfb.ac.cn/ calculation/pp.htm, and was used to analyze the relationship with protein purification.
.However it appears 4 times in this protein, so the amino acid pair TT is unpredictable. By this way, all amino acid pairs can be classified as predictable or unpredictable, and their sums constitute the predictable and unpredictable portions of a protein. For this protein, its predictable and unpredictable portions are 55.25% and 44.75%. This feature is generally different from protein to protein, which can be computed at the web https://www.nerc-nfb.ac.cn/ calculation/pp.htm, and was used to analyze the relationship with protein purification.
Modeling
It is necessary to compare the relationship between each constant or varying feature of amino acids (model inputs) and the purification state (model output) [14-19] in order to find out which feature can give the best prediction. This was modeled using MatLab with both logistic regression and 10-1 feed-forward back-propagation neural network [28,29].
Statistics
The results were classified into true positive, false positive, true negative and false negative. The accuracy, sensitivity and specificity were calculated as follows:
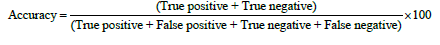
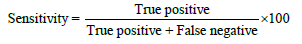
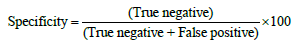
The data were presented as median with inter-quatile, and were analyzed by Chi-square test. Kruskal-Wallis one way ANOVA on ranks and Mann-Whitney rank sum test were used to analyze the difference among and between different predictions. The receiver operating characteristic (ROC) analysis was used to compare the sensitivity and specificity [30-32]. P < 0.05 was considered statistically significant.
Figure 1 is the heat map of the results using logistic regression to model the relationship between the success rate of purification of S. cerevisiae proteins and each of 535 amino acid features. In this figure, the y-axis indicated each of 535 amino acid features used as a predictor, and the x-axis indicated the accuracy, sensitivity and specificity of prediction results. Two characters can be drawn from Figure 1:

Figure 1: Heat map of accuracy, sensitivity and specificity obtained from modeling the relationship between the success rate of purification of S. cerevisiae proteins and each of 535 amino acid features using logistic regression.
(1) The prediction provides very high sensitivity with relative low specificity;
(2) Different amino acid features give similar results (rows 3-6 in Table 2) and especially most of constant amino acid features provide the same prediction results indicated by the same color.
| Amino acid feature Logistic regression | Number | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|
| Constant composition features | 40 | 0.735 (0.735 - 0.735) [0.713 - 0.735] | 0.929 (0.929 - 0.929) [0.913 - 0.929] | 0.337 (0.337 - 0.337) [0.304 - 0.337] |
| Physicochemical features | 218 | 0.735 (0.735 - 0.735) [0.664 - 0.741] | 0.929 (0.929 - 0.929) [0.919 - 0.949] | 0.337 (0.337 - 0.337) [0.0873 - 0.342] |
| Second structure features | 273 | 0.735 (0.735 - 0.735) [0.697 - 0.741] | 0.929 (0.929 - 0.929) [0.922 - 0.938] | 0.337 (0.337 - 0.337) [0.205 - 0.354] |
| Dynamic features Modeling | 4 | 0.676 (0.668 - 0.708) a,d [0.666 - 0.735] | 0.93 (0.905 - 0.934) [0.882 - 0.936] | 0.198 (0.124 - 0.303) d,f [0.12 - 0.337] |
| Constant composition features | 40 | 0.696 (0.679 - 0.704) [0.672 - 0.782] | 0.967 (0.952 - 0.985) [0.896 - 1] | 0.142 (0.0455 - 0.198) [0 - 0.484] |
| Physicochemical features | 218 | 0.724 (0.672 - 0.773) a [0.672 - 0.781] | 0.932 (0.927 - 0.997) a [0.865 - 1] | 0.359 (0.012 - 0.456) [0 - 0.486] |
| Second structure features | 273 | 0.77 (0.764 - 0.774) a,d [0.672 - 0.781] | 0.925 (0.919 - 0.93) c,e [0.879 - 1] | 0.458 (0.444 - 0.466) c,e [0 - 0.498] |
| Dynamic features | 4 | 0.764 (0.723 - 0.78) [0.692 - 0.785] | 0.919 (0.909 - 0.936) b,d [0.905 - 0.948] | 0.447 (0.297 - 0.504) a [0.168 - 0.539] |
| Validation Constant composition features | 40 | 0.687 (0.676 - 0.691) [0.672 - 0.727] | 0.954 (0.938 - 0.984) [0.867 - 1] | 0.138 (0.0435 - 0.18) [0 - 0.393] |
| Physicochemical features | 218 | 0.698 (0.673 - 0.724) a [0.671 - 0.731] | 0.899 (0.888 - 0.997) a [0.852 - 1] | 0.308 (0.0059 - 0.387) a [0 - 0.397] |
| Second structure features | 273 | 0.722 (0.718 - 0.725) c,e [0.672 - 0.73] | 0.886 (0.884 - 0.893) c,e [0.86 - 1] | 0.388 (0.384 - 0.391) c,e [0 - 0.397] |
| Dynamic features | 4 | 0.689 (0.676 - 0.71) f [0.671 - 0.723] | 0.875 (0.842 - 0.912) b,d [0.818 - 0.94] | 0.357 (0.249 - 0.38) [0.151 - 0.39] |
The data are presented as median with inter-quatile in parentheses and range in brackets. The letters of a, b and c indicate statistical significance at P<0.05, P<0.01 and P<0.001 levels, respectively, compared with constant composition features (Mann-Whitney Rank Sum Test). The letters of d and e indicate statistical significance at P<0.05 and P<0.001 levels compared with physicochemical features (Mann-Whitney Rank Sum Test). The letter f indicates statistical significance at P<0.05 level compared with second structure features (Mann-Whitney Rank Sum Test).
Table 2: Results obtained from logistic regression, modeling and delete-1 jack-knife validation by means of 10-1 feed-forward back-propagation neural network.
These two characters reveal obvious in Table 2 that lists the statistic results of predictive performance grouped by the model inputs and the amino acid features.
Figure 2 is the heat map of the results using 10-1 feedforward backpropagation neural network to model the relationship between the success rate of purification of S. cerevisiae proteins and each of 535 amino acid features (left 3 columns), and to valid this relationship with delete-1 jackknife validation (right 3 columns). Compared with logistic regression, neural network significantly enhances the ability of screening of model inputs because different amino acid features generate different prediction results as seen different colors in Figure 2 and numerical comparison in both modeling (rows 8-11 in Table 2) and validation (rows 13-16 in Table 2).

Figure 2: Heat map of accuracy, sensitivity and specificity obtained from modeling the relationship between the success rate of purification of S. cerevisiae proteins and each of 535 amino acid features using 10-1 neural network, and from validating this relationship with delete-1 jack-knife validation.
Figure 3 displays the ROC analysis with the aim to furthermore distinguish the performance of amino acid features as predictors in logistic regression (topper panel), in neural network modeling (middle panel) and delete-1 jackknife validation (bottom panel). Clearly, all predicted results were located in the up-left triangle, indicating that both models are effective to predict the success rate of purification of S. cerevisiae proteins because the predictions surpass a random guess, which is the diagonal line. Some amino acid features have better predictive performance with high sensitivity and relatively high specificity as marked in pink circle.

Figure 3: ROC analysis for sensitivity and specificity obtained from modeling the relationship between the success rate of purification of S. cerevisiae proteins and each of 535 amino acid features. The diagonal line is the line of indiscrimination indicating a completely random guess.
Figure 4 illustrates the predictive results in each S. cerevisiae protein by neural network. In this figure, the x-axis of upper and middle panels represents 1294 S. cerevisiae proteins, which are ranked according to their predictive accuracy. The upper panel represents the predictive accuracy obtained from modeling (green bars) while the middle panel represents the predictive accuracy obtained from delete-1 jackknife validation (light blue bars). In this way, S. cerevisiae proteins are divided into two groups by the cut-off point of 90% accuracy that is set as an acceptable accuracy. The lower panel of Figure 4 shows the statistical difference between the two groups in terms of predictable portion of S. cerevisiae portions. As can be seen, the larger the predictable portion is, the better the prediction is.

Figure 4: Accuracy of purification of S. cerevisiae proteins obtained from modeling (upper panel) and delete-1 jack-knife validation (middle panel), and statistical comparison of their predictable portion of amino acid pairs (lower panel). The dotted lines indicate the cut-off point for separating the low accuracy from the high one. The data were presented as median with interquartile. [*indicates the statistically significant difference compared with the group of low accuracy at P<0.001 level (Mann-Whitney Rank Sum Test)].
This study focuses on analyzing the success rate of purification of S. cerevisiae proteins through modeling for the purpose of finding out a simple way to predict the purification state of a protein. Both logistic regression and neural network can be used to model the relationship between an amino acid feature and the purification state. However, the results demonstrate that logistic regression is weak in screening of different inputs whereas neural network reveals more powerful to distinguish the predictive performance of different amino acid features. This is consistent with previous studies for predicting crystallization propensity of proteins [14-19]. The results confirm that some amino acid features can serve as predictors to predict the success rate of protein purification. The predicted performance for S. cerevisiae proteins shows a very high sensitivity accompanied by a relatively low specificity, thus searching a suitable predictor should be focus on the outcome of specificity. Accordingly, the varying features are considered to be best predictors because 3 out of 4 of them provide better results as indicated in the pink circles in Figure 3. Three varying features are used in this study: future composition of amino acids, distribution probability of amino acids and amino acid pare predictability. These features are varying in different proteins with different amino acid composition, position and neighboring amino acid, and reveal their benefit for analyzing protein structure and function in many ways [25,26].
Purification is one of the most significant cost steps for producing recombinant proteins. Many efforts have been made to improve purification process in order to produce proteins more efficiently [33]. Some methods have been established for predicting multi-step experimental procedures of protein production including purification. For example, PredPPCrys is a new approach using the support vector machine and a comprehensive set of multifaceted sequence-derived features in combination with a novel multi-step feature selection strategy [34]. In comparison, this study shows a very simple method to predict the success rate of protein purification based on its sequence information, which can give researchers a concept what a chance a protein would be purified before conducting experiment. Interestingly, the simplest varying feature such as the amino acid composition of a protein can be used to predict the success rate of protein purification.In this way, the predictive process is quite simple so that one can easily have a concept about the probability a protein to be purified, which should be helpful to overcome blindfold experiments and enhance the production of designed proteins.
This study analyzes the relationship between the success rate of purification of S. cerevisiae proteins and each of 535 amino acid features through modeling. The results demonstrate that the predicted performance of neural network is more powerful than that of logistic regression. Some amino acid features can be used as predictor to predict the purification tendency of proteins, and among them the variable amino acid features show better predictive outcomes. In general, the sensitivity is very high while the specificity is low. Also, the S. cerevisiae proteins with high predictable portion of amino acid pairs will have higher accuracy of purification prediction than those with low predictable portion.
Table S1. List of 1294 proteins from S. cerevisiae used in this study.
Table S2. List of 535 features of amino acids used in this study.
Table S3. Amino acids and their translated amino acids with translation probability.
This study was partly supported by National Natural Science Foundation of China (31460296 and 31560315), Guangxi Natural Science Foundation (2013GXNSFDA019007), Guangxi Scientific and Technological Cooperation and Exchange Program (1347004-1) and Special Funds for Building of Guangxi Talent Highland.