Journal of Clinical and Experimental Ophthalmology
Open Access
ISSN: 2155-9570
ISSN: 2155-9570
Research Article - (2019) Volume 10, Issue 3
Unutilized, enormous intracellular glucose in insulin-nondependent tissues, including retina, leads to several consequences: (i) increased formation of advanced glycation end products, (ii) activation of polyol pathway, (iii) anaerobic glycolysis, (iv) glutamate toxicity, (v) lipid peroxidation and oxidative stress, and all of these, finally, resulting in convergence to up regulation of antigenic VEGF and VEGFR2, the crucial player in the development of DR. The purpose of the present pilot study is to assess the effects of one kind of intervention on the development of DR: supplementation of B-vitamins (B1, B2, B3, B5 and B6), vitamin C and vitamin E on amelioration of biochemical derangements related to development of DR.
400 diagnosed type 2 diabetic patients were identified for the study, of which subjects were randomised 1:1 to receive oral antidiabetic medication along with B-vitamins, vitamin C and vitamin E or only antidiabetic medication to give the study and controlled population in this unblended randomized trial since December 2004 to December 2017. The following preliminary tasks were completed: firstly, baseline detailed fundoscopic examinations were enough to exclude the presence of retinopathy. Secondly, had baseline biochemical parameters like bllod concentration of NAD+, NADH, advanced glycation end products (AGEs), malondialdehyde (MDA), VEGF and VEGFR2 determined? Lastly, yearly fundoscopic examinations were documented to detect the features of DR.
These efforts revealed the following findings: 32 among 187 (17.11%) patients who received supplementation with B-vitamins, vitamin C and vitamin E developed very mild microangiopathy; whereas 92 patients among 200 (46%) controlled ones developed mild to moderate non proliferative diabetic retinopathy (NPDR). 13 patients of the study group who didn’t follow up were counted as lost from the observation of the study.
The findings lead to this conclusion: Glycolysis and citric acid cycle should run uninterruptedly by supplementation of precursors of oxidized cofactors and antioxidants to prevent biochemical derangements which lead to increased expression of VEGF.
Keywords: Type 2 Diabetes Mellitus (T2DM); Diabetic Retinopathy (DR); Vitamin B; Vitamin C; Vitamin E; Biochemical derangement; Nicotinamide Adenine Dinucleotide Positive (NAD+), Reduced Nicotinamide Adenine Dinucleotide (NADH); Red Blood Cell (RBC)
In spite of remarkable advances in diagnosis and treatment, diabetes mellitus (DM) is becoming rampant and assuming the proportions of a global epidemic,with its most common microvascular complication, diabetic retinopathy, rising as the leading cause of blindness among the working age groups [1]. Duration and severity of hyperglycemia have been identified as the major contributors to the pathogenesis of DR [2]. So far as the explicable etiopathogenesis known till date is concerned, it ’ s found that hyperglycemia-induced toxic metabolic pathways are considered to lead to degeneration of microvasculature of retina and gradual vaso-occlusion of retinal capillary bed resulting in an ischemic environment and tissue hypoxia. Though there are many mechanisms implicated in pathogenesis of microangiopathy of DM, the background mostly explored tells that the initiation of inflammation is the convergence of the following symptoms: environment of hyperglycemia, abnormal lipids, increased oxidative stress, elevated serum and tissue advanced glycation end products, and increased serum and tissue cytokines. Pathways of inflammation incite the pathways of endothelial dysfunction and vaso-occlusion of retinal capillaries resulting in tissue hypoxia which invite increased secretion of vascular endothelial growth factor [3].
In the last decade, many of the investigators in this field were attracted by the flames of inflammation, which is established as the principal factor related to the causation of diabetic retinopathy. Sequential experimental animal and human studies demonstrated supportive evidences for this inflammatory etiology-mediated microangiopathy [4-6].
Explicable biochemical scenario and persistent replicative observations, including the one done by us, postulated that vascular endothelial growth factor (VEGF) is the crucial mediator for DR [7,8]. Therefore the scenario of promising therapeutic efficacy changed the treatment paradigm of DR from painful laser photocoagulation to pharmacotherapy with anti-VEGF agents that demonstrated a significant progress made in large proportions of diabetic patients with vision impairing diabetic retinopathy [9]. At the same time, the other unavoidable fact is that repeated intravitreal injections of anti-VEGF agents and maximal anti-inflammatory therapy cannot ameliorate blindness owing to diabetic macular edema and neovascularization of diabetic retinopathy in a significant portion of the diabetic population [10]. This perspective of the present therapy draws our attention towards other factors related to causation of tissue hypoxia, the principal mechanism of development and progression of DR. The red blood cells (RBCs) which carry oxygen to tissue level play a remarkable role in causation of the disease owing to their structural and functional alterations behind the biochemical and inflammatory background of DR. In this present study, we also attempted to prevent the detrimental effects of hyperglycemia-induced biochemical derangements and the structural and functional abnormalities of RBCs related to production of tissue hypoxia.
Subjects
A total count of 400 patients of type 2 DM were recruited following informed consent under the declarations of Helsinki. Sample size was calculated for each group as per formula proposed by Naing et al. [11].
The formula is:
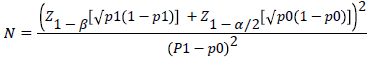
Where; P1is the anticipated proportion=40%
Po is the Null Hypothesis value or actual proportion of progression of diabetic subjects to DR within 15 years=53% [The Wisconsin Epidemiologic Study of diabetic retinopathy, 1984].
(p1-p0) is the difference between proportions=0.13
Z 1-β is the desired power of study=90% (1.28)
Z 1-α/2 is the desired level of significance=5% (1.96)
Alternate hypothesis is two sided
Putting the values in the formula the required sample size in each group is: 152
30% subject may loss to follow up in each group=46
So, total sample size in each group=152+46=198 ≈ 200 (taking round up value).
Patients who were primarily diagnosed in the diabetic clinic of the institute of Post-graduate Medical Education and Research, Kolkata, India, according to WHO guidelines and criteria of American Diabetic Association, and referred to the Retina Research Clinic of the Regional Institute of Ophthalmology, Kolkata, India, were recruited for comprehensive ophthalmological examination during the period from December 2004 to December 2005.
Inclusion criteria were (1) diagnosis of type 2 DM of ≤ 5 years; (2) Stable medication with oral hypoglycemic agents for the management of diabetes before entry into the study and expected to remain during the study; (3) best-corrected visual acuity ≥ 6/9 in each eye with no DR; and (4) literate patients who had the ability to give informed written consent and could read the Snellen’s visual acuity chart.
The exclusion criteria were (1) presence of DR with or without diabetic macular edema as determined by dilated fundoscopic examination, spectral domain optical coherence tomography (SDOCT), and digital fundus photography; (2) patients taking insulin or history of taking insulin; (3) concomitant conditions like branch retinal vein occlusion, Eales’ disease or presence of glaucoma in the eyes that could alter the normal occurrence of microvascular complication of the disease; (4) patients suffering from hypertension, coronary artery diseases or strong family history of coronary artery diseases, peripheral vascular disease, chronic infection, thromboembolic events, urinary microalbumin>300 mg/dl were some of the conditions considered as exclusion criteria of this study.
Treatment protocol
The study subjects who were diagnosed by an experienced retinologist to have no retinopathy at baseline were given standard doses of the following: (i) vitamin B containing thiamin mononitrate 10 mg, (ii) riboflavin 10 mg, (iii) nicotinamide 45 mg, (iv) pyridoxamine HCl 3 mg, (v) Ca panthenate 50 mg, (vi) cyanocobalomine 15mcg (Neurobionforte, MERK PHARMA); (vii) ascorbic acid 500 mg (Celin, Glaxosmithkline Pharmaceuticals ); and lastly,α-tocopherol 400 (Evion, MERK LTD India) along with required oral hypoglycemic agents. General physical health and ocular condition were assessed at baseline and at a 12 months interval using standard clinical procedures for evaluating vision and ocular pathology, including, (i) best corrected visual acuity by Snellen’s Visual Acuity Chart, (ii) intraocular pressure by applanation tonometry, (iii) contrast sensitivity by Robin Contrast Sensitivity Chart, (iv) dilated fundus examination by direct, indirect ophthalmoscopy and slit-lamp biomicroscopy with + 90D lens, and (v) central retinal thickness measurement by spectral-domain optical coherence tomography (Spectralis, Heidelberg Engineering, Germany).
Efficacy and safety assessment
General health of each subject was assessed during the course of the study, including blood pressure, heart rate, body weight, HbA1c readings and urinary microalbumin/creatinine ratio. Subjects who exhibited sensitivity to any of the orally administered vitamins were noted and excluded from the efficacy assessments.
Laboratory investigations
Duly signed by the participants, written informed consent was taken before the following investigations were carried out at the base-line and at the end of 12 years of the follow-up study. First, the collection of 15 ml of venous blood by venepuncture from all the recruited subjects was done after theirs having undergone a fasting period of 12 hours duration. From these, 10 ml blood samples were collected in a heparinized tube. From 10 ml heparinized blood sample, 8 ml was collected in a 15 ml sterile centrifuge for peripheral blood mononuclear cell (PBMC) isolation, and 2 ml samples were centrifuged at 3000 revolutions per minute for 10 minutes at 4°C to separate cellular components and plasma. Buffy coat was removed by careful aspiration, and packed erythrocytes were then washed with cold phosphate buffered saline (pH-7.2) by maintaining 4°C temperature for estimation of erythrocytes redox state. Plasma samples were collected in cryocube vials for assay of glucose. The remaining 5 ml sample from the initial 15 ml sample was aliquoted in to two vials. 3 ml in a clot vial to obtain serum for estimation of AGE, MDA and VEGF.2 ml sequestrenated whole blood was used for morphological study of red blood cells.
Measurement of erythrocyte pyridine coenzymes
Buffy coat was removed from centrifuged blood by careful aspiration, and packed erythrocytes were washed with cold phosphatebuffered saline (pH 7.2) at 4°C and 106 red blood corpuscle (RBC) cells were pelleted in microcentrifuge tube (200 revolutions per minute for 5 minutes ) for the estimation of coenzymes ( NAD+, NADH). The concentration of pyridine coenzymes and their ratio were determined by using the kits of Bio Vision (Bio Vision Research Products, Mountain View, CA). Nicotinamide adenine dinucleotide positive and NADH concentrations in erythrocytes were measured by NAD cycling enzyme mixture through an enzyme cycling reaction. The determinations included both the oxidized and reduced form of nucleotide. The absorbance of color product was read at 450 nm by Bio-Rad multiplate reader (Model 680). Furthermore, NAD+: NADH ratio was calculated as follows:
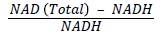
Measurement of peripheral blood mononuclear cell reactive oxygen species
Mononuclear cells were obtained from 8 ml heparinized blood by using Histopaque-1077 density gradient separating media (Sigma Aldrich, St. Louis, MO, USA) for 40 minutes at 1300 rpm and 20°C. PBMCs were further subjected to centrifugation at 1500 rpm for 10 minutes and washed with phosphate-buffered saline twice. Cells (5 × 105)were pelleted and resuspended in phosphate-buffered saline for the estimation of ROS. Intracellular ROS generation in mononuclear cells was measured by ROS sensitive cell permeable dye 2’7’dihydro dichlorofluorescein diacetate (2’7’H2DCF-DA), which in the presence of ROS was oxidized to highly fluorescent 2’7’-dichlorofluorescein (2’7’DCF) in the cell. Production of intracellular ROS is directly proportional to the oxidation of 2’7’H2DCF-DA and thereby elevates cellular fluorescence level. 5 × 105 number of pelleted cells were washed twice with IX PBS (pH 7.2) by centrifuging at 4000 rpm for 5 minutes and cells were resuspended in 500 μL of IX PBS (pH 7.2). Thereafter cells were incubated with 20 μm 2’7’H2DCF-DA for 30 minutes at 37°C. finally the cells were washed again with IX PBS (pH 7.2) and resuspended in 400 μL of IX PBS. The mononuclear cells exhibiting increased fluorescence of oxidized DCF was measured by flow cytometry ( FACS caliber, Becton Dickinson, San Jose, CA) equipped with an argon ion laser (15 mW) tuned to 488 nm. The fluorescence of DCF was collected in FL1 channel, equipped with a 530/30 nm band pass filter. Fluorescence was measured in the long mode using cell quest prosoftware (BD Bioscience, San Jose, CA) and expressed as geometrical mean fluorescence channel (GMFC). Cells were gated on the basis of their characteristic morphology i.e. forward scatter and side scatter of monocytes and lymphocytes. Acquisitions were performed on 10000 gated events; while data analysis was carried out with cell questpro software (BD Bioscience).
Measurement of MDA
The quantitative analysis of serum malondialdehyde (MDA) free of interference from sialic acid was used as described by Satoh [12]. Lipoproteins in serum were precipitated by adding 20% trichloroacetic acid and 8.1% dodecyl sulphate. Thereafter, 0.8% aqueous solution of thiobarbituric acid was added to this precipitate mixed well and finally was heated at 95°C for 1 hour for coupling of lipid peroxide with thiobarbituric acid reagent. The resulting chromogen was extracted from the precipitate by adding a mixture of n-butanol and pyridine (15:1). The organic mixture was separated by centrifugation and the intensity of the organic layer was measured spectrophotometrically (Halo DB-20, Dynamica, Salzburg-Mayrwise, Austria) by using a 530 nm filter against water blank. The concentration of MDA in serum sample was determined from a linear standard curve established by 1 to 8 nm of 1,1,3,3 tetra methoxy propane.
Measurement of advanced glycation end-products (AGEs) from serum
Serum level of total advanced glycation end products was measured by enzyme-linked immunosorbent assay by using The Cell Biolabs kit (Cell Biolabs, San Diego, CA). AGE protein adducts present in the sample were probed with an ant-AGE polyclonal antibody, followed by HRP conjugated antibody. The AGE-protein adduct content in the sample was determined by comparison with a standard curve prepared from AGE-bovine serum albumin (BSA) standards ranging from 0.25 to 5 μg/ml. The absorbance of the final color product was read at 450 nm as the primary wavelength using a Bio Rad multiplate reader (Model 680, Bio Rad, Laboratories, Hercules, CA) against the reduced BSA standard as the absorbance blank. The AGE-BSA provided in the kit was prepared by reacting BSA with glycolaldehyde, followed by extensive dialysis and column purification.
Measurement of erythrocytes 2, 3-DPG content
The erythrocytes 2, 3-DPG content was determined by quantitative sandwich enzyme immunoassay technique using the Cusabio Human 2, 3-DPG ELISA kit (CSB-E09861h, CUSABIO, China). At first hemolysate of erythrocytes was prepared as per method described by Erlandsen et al. [13]. Then hemolysate was used in the assay. Briefly, Standards and samples were pipetted into the 2, 3-DPG specific antibody coated wells. Then any 2, 3-DPG present in the sample or standard was bound by the immobilized antibody. After removing any unbound substances, a biotin-conjugated antibody specific for 2, 3- DPG was added to the wells. After washing, avidin conjugated Horseradish Peroxidase (HRP) was added to the wells. Following a wash to remove any unbound avidin-enzyme reagent, a substrate solution was added to the wells and color developed in proportion to the amount of 2, 3-DPG bound in the initial step. The color development was stopped by addition of stop solution and the intensity of the color was measured colorimetrically by using 450 nm filter in an ELISA plate reader (BIO-Rad, Model 680).
Erythrocyte osmotic fragility study
Osmotic Fragility of RBC was tested as per modified method of Parpart et al. [14]. In brief, 50 μl of heparinized blood sample was pipetted in to a set of tubes containing 5 ml of each 0.1, 0.3, 0.5, 0.7, 0.9 gm/l of NaCl (pH 7.4) and there after carefully mixed and incubated for 30 minutes at room temperature (26°C-28°C). Then the test tubes were centrifuged at 1500 × g for 5 minutes. The supernatant was transferred in to a glass cuvette and the absorbance was read spectrophotometrically at a wave length 415 nm. The percentage of haemolysis for each concentration of NaCl was calculated using the formula below-
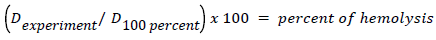
Where, the optical density, D equals the log of the reciprocal of transmission value obtained in the colorimeter, D 100 percent is the calibration solution, D experiment, is that for the supernatant fluid of any particular blood-salt solution. The degree of hemolysis was calculated by comparing with 0.1% NaCl which represent 100% lysis.
Scanning electron microscopy of Red Blood Cells (RBCs)
Red cells were prepared for SEM examination by fixing the sequestrenated whole blood in 2.5% buffered glutraldehyde prepared in 2 molar cacodylate buffers for one and half hour for room temperature for primary fixation. Red blood cells were separated from white blood cells by centrifugation. After primary fixation, the cells were washed with 1 molar cacodylate rinsing buffer three times for 15 minutes at room temperature. After washing, the samples were fixed in 1% osmium tetraoxide for one hour for secondary fixation. The postfixed cells were washed in 0.1 molar cacodylate rinsing buffer and in distilled water three times for 15 minutes at room temperature.
After secondary fixation, the cells were dehydrated in graded series of ethanol. The samples were vaccum dried in dessicator for overnight. After drying, samples were mounted on aluminium stubs with double adhesive carbon tapes and coated with gold for 20 seconds in a gold ion sputter to render their surfaces conducting. Red cells were visualized and photographed in a Scanning Electron Microscope (Product No: EVOIS-20-67, EVO 18, EVO/18, Carlzeiss, Germany). Microphotographs were taken at direct magnification in 5 KX.
Measurement of surface roughness of RBC
The surface roughness of Scanning electron microscopic image of an RBC was performed by using ‘Image J’ software (version 1.46, Rasband, NIH). First, a randomly selected scanning electron microscopic image of an RBC from a film was converted to 32-bit. Then 3D surface presentation and gradient analysis of the 3D surface was done by using ‘Plugins’. The values of RA (roughness average), RQ (root mean square roughness), Rsk ( skewness of the roughness profile) and Rku ( kurtosis of the roughness profile) were considered as the determinant of the surface roughness for the present study.
At baseline red cells were prepared from 48 blood samples of the control group and 47 blood samples of the study subjects. Six individuals of the study group were lost to follow-up. So ultimately, red blood cells of 48 control population and 41 study subjects were again analyzed by the same procedures of Scanning Electron Microscopic study at the end point of 12 years follow-up and relevant data were entered into statistical evaluation.
Measurement of serum Vascular Endothelial Growth Factor (VEGF)
Serum VEGF was assayed by ELISA using commercial kits of Ray Biotech (Ray Biotech, Norcross, USA). In this assay, an antibody specific for human VEGF was coated on a well plate. A series of standards ranging from 8.23 pg/ml to 6000 pg/ml and samples (serum samples were two fold diluted with assay diluents) were added into the wells. VEGF protein present in the sample was bound to the wells by the immobilized antibody. The wells were washed and biotinylated antihuman VEGF antibody was added. After buffer wash HRPconjugated streptavidin was pipette into the wells. Further, the wells were subjected to wash and TMB substrate solution was added into the wells and was placed in incubation at room temperature for 30 minutes. Intensity of the final color product was proportional to the concentration of VEGF protein present into the samples and the absorbance of the color product was measured colorimetrically by using 450 nm filters in the ELISA plate reader (Bio Rad, Model 680). The concentration of VEGF in the samples was determined by a standard curve and the assay detected less than 10 pg/ml of VEGF from the sample.
The procedures of estimation of VEGF and surrogate markers of oxidative stress, lipid peroxidation and advanced glycation end product formation had been also previously described in our previous works [15-17].
Measurement of plasma glucose
Plasma glucose levels were measured by glucose oxidase and peroxidase method. The absorbance was read at 520 nm in a spectrophotometer Halo DB-20; Dynamica).
Measurement of glycosylated hemoglobin
Glycosylated hemoglobin was measured by direct enzymatic assay, using the kit of Diazyme laboratories (Poway, CA). The assay involved extensive protease digestion of lysed whole blood samples with Bacillus species protease. The concentration of glycosylated hemoglobin was expressed as hemoglobin A1 c% and measured by use of a calibration curve in which the calibrators have values for each level of hemoglobin A1c%. The assay has a linear range from 4.0% to 16.0% and was performed in biochemistry autoanalyser (Hitachi 980).
Repeatability and durability of each biochemistry analysis were maintained by matching the values of control with repeat analysis on the same day and over a period of 20 days.
We have maintained internal quality control by calibrating the machines regularly, running the samples in duplicate and comparing the values between the study and control groups. All the biochemical procedures was done by one of the investigators to reduce the inter person human error.
Statistical analysis
Data were expressed as mean ± SE (standard error of means). Normally distributed data were evaluated by paired ‘ t ’ test and unpaired‘t’ test respectively. Not normally distributed paired data were evaluated by Wilcoxon signed rank test and unpaired data were evaluated by Mann-Whitney U test. Spearman correlation was performed to find out the correlation between two not-normally distributed parameters. Statistical analysis for gender distributions was evaluated by chi-square test using Graph pad prism software (Version 6, Sandiego, CA, USA). Kaplan-Meier survival analysis was performed to determine the probability of survival from DR among diabetic individuals of group-A and B respectively. The rate of the study groups was further compared with Log-rank test (Mantel-Cox) by using IBM SPSS software (Version 25, SPSS, Inc, and Chicago, USA). Values of p<0.05 were considered to be minimum level of significance.
Baseline clinical characteristics of study subjects and controls show no significant differences in age, gender, duration of diabetes, body mass index, blood pressure, plasma glucose, glycosylated hemoglobin and plasma lipid levels (Table 1).
| Parameters | Particulars | Group-A N=200 | Group-B N=200 | p-value |
|---|---|---|---|---|
| Age(years) | 53.55 ± 0.79 | 5.48 ± 0.98 | 0.402 | |
| Gender | Male | 110 (55%) | 116 (58%) | 0.545 |
| Female | 90 (45%) | 84 (42%) | ||
| BMI (Kg/m2) | 24.40 ± 0.34 | 23.57 ± 0.48 | 0.161 | |
| BP (mm Hg) | Systolic | 127.2 ± 0.98 | 126.4 ± 0.90 | 0.529 |
| Diastolic | 80.58 ± 0.70 | 81.16 ± 0.69 | 0.569 | |
Table 1: Baseline demographic and physiological parameters of the study groups.
Comparison of group-A and group-B in the present study showed no statistically significant differences for baseline biochemical parameters like FPG, PPG, HbA1c, ROS, MDA, AGE, NAD+, NADH, NAD+/NADH, 2,3-DPG and VEGF. Comparison of same biochemical parameters of group–A at baseline with group-A after 6 and 12 years also showed no statistically significant differences. However, the biochemical parameters like ROS, MDA, AGE, NADH and VEGF were found to be significantly higher in group-B after 6 and 12 years in comparison to their baseline levels. The study also showed lower levels of NAD+, NAD+/NADH and 2, 3-DPG in group-B after 6 and 12 years than their baseline levels. Regarding glycemic parameters like FPG, PPG and HbA1c, no statistically significant differences were observed between group-A and group-B after 6 and 12 years. However, group-B after 6 and 12 years showed higher levels of ROS, MDA, AGE, NADH and VEGF and lower levels of NAD+, NAD+/NADH and 2,3-DPG in comparison to group-A (Tables 2-4). There was a trend of incremental changes of biochemical parameters (ROS, MDA, AGE and VEGF ) observed over time.
| Parameters | Particulars | Group-A (Baseline) N=200 | Group-B (Baseline) N=200 | Group A vs. Group B (Baseline) p-value | Group-A (After 12 years) N=187 | Group-B (After 12 years) N=200 | Group A vs. Group B (after 12 years) p-value | Group-A baseline vs. Group-A (after 12 years) p-value | Group-B baseline vs. Group-B (after 12 years) |
|---|---|---|---|---|---|---|---|---|---|
| Plasma glucose (mg/dl) | FPG | 150.7 ± 1.86 | 154.6 ± 2.21 | 0.213 | 149.5 ± 1.77 | 153.3 ± 3.01 | 0.179 | 0.628 | 0.842 |
| PPG | 191 ± 5.03 | 194.5 ± 5.22 | 0.511 | 189.6 ± 4.82 | 204 ± 10.94 | 0.128 | 0.788 | 0.332 | |
| HbA1c % | 7.86 ± 0.14 | 7.56 ± 0.14 | 0.15 | 7.98 ± 0.15 | 7.71 ± 0.24 | 0.359 | 0.385 | 0.452 | |
| ROS (geomean of DCF/5×105 cells) | 96.31 ± 0.90 | 96.18 ± 0.66 | 0.812 | 98.80 ± 2.33 | 112.6 ± 1.63 | 0.0002 | 0.642 | ˂0.0001 | |
| MDA (nmol/ml) | 2.69 ± 0.03 | 2.63 ± 0.03 | 0.497 | 2.76 ± 0.13 | 3.24 ± 0.08 | 0.038 | 0.769 | ˂0.0001 | |
| AGE (µg/ml) | 2.55 ± 0.10 | 2.65 ± 0.05 | 0.38 | 2.67 ± 0.13 | 3.59 ± 0.23 | 0.002 | 0.791 | ˂0.0001 | |
| NAD+(pmol/106 RBC) | 47.19 ± 0.77 | 47.71 ± 01.49 | 0.322 | 45.29 ± 1.03 | 39.92 ± 1.26 | 0.003 | 0.231 | 0.0007 | |
| NADH (pmol/106 RBC) | 4.42 ± 0.16 | 3.95 ± 0.25 | 0.225 | 4.52 ± 0.10 | 5.56 ± 0.31 | 0.003 | 0.571 | 0.003 | |
| NAD+/NADH | 10.88±0.44 | 12.98±0.92 | 0.152 | 10.5±0.22 | 7.64±0.52 | ˂0.0001 | 0.133 | ˂0.0001 | |
| 2,3-DPG (µmol/g Hb | 17.17 ± 0.30 | 17.48 ± 0.54 | 0.615 | 16.20 ± 0.72 | 13.42 ± 0.68 | 0.012 | 0.131 | 0.0002 | |
| VEGF (pg/ml) | 92.33 ± 1.23 | 93.07 ± 1.75 | 0.722 | 94.18 ± 2.90 | 131.7 ± 4.05 | <0.0001 | 0.804 | ˂0.0001 |
Table 2: Biochemical parameters of Group-A and Group-B at the time of diagnosis of T2DM and 12 years after treatment of Group-A (with antihyperglycemic drugs, B-vitamins, ascorbic acid and α-tocopherol) and group-B (only with anti-hyperglycemic drugs).
| Parameters | Particulars | Group-A (Baseline) N=200 | Group-B (Baseline) N=200 | Group A vs. Group B (Baseline) p-value | Group-A (After 12 years) N=187 | Group-B (After 12 years) N=200 | Group A vs. Group B (after 12 years) p-value | Group-A baseline vs. Group-A (after 12 years) p-value | Group-B baseline vs. Group-B (after 12 years) p-value |
|---|---|---|---|---|---|---|---|---|---|
| RQ | 35.95 ± 1.69 | 37.04 ± 1.70 | 0.655 | 36.90 ± 1.61 | 44.17 ± 1.70 | 0.006 | 0.355 | 0.01 | |
| RA | 25.42 ± 1.32 | 25.73 ± 1.34 | 0.86 | 26.10 ± 1.32 | 31.22 ± 1.58 | 0.021 | 0.219 | 0.018 | |
| Osmotic Fragility | Starting | 5.44 ± 0.09 | 5.37 ± 0.08 | 0.586 | 5.60 ± 0.07 | 6.07 ± 0.07 | 0.0002 | 0.189 | <0.0001 |
| (conc. Of NaCl) | Complete | 4.44 ± 0.11 | 4.41 ± 0.15 | 0.856 | 4.69 ± 0.14 | 5.12 ± 0.14 | 0.047 | 0.243 | 0.003 |
| CMT (µm) | 244.4 ± 1.56 | 246 ± 1.48 | 0.297 | 250.1 ± 4.41 | 255.6 ± 7.91 | 0.278 | 0.812 | 0.131 | |
| VA (log MAR) | 0.22 ± 0.01 | 0.23 ± 0.01 | 0.669 | 0.245 ± 0.02 | 0.34 ± 0.03 | 0.04 | 0.319 | 0.004 | |
Table 3: Red blood corpuscles surface roughness, osmotic fragility, retinal structural and functional parameters of Group-A and Group-B at the time of diagnosis of T2DM and 12 years after treatment of Group-A (with anti-hyperglycemic drugs, B-vitamins, ascorbic acid and α-tocopherol) and group-B (only with anti-hyperglycemic drugs).
| Parameters | Particulars | Group-A (Baseline) N=200 | Group-B (Baseline) N=200 | Group A vs. Group B (Baseline) p-value | Group-A (After 12 years) N=187 | Group-B (After 12 years) N=200 | Group A vs. Group B (after 12 years) p-value | Group-A baseline vs. Group-A (after 12 years) p-value | Group-B baseline vs. Group-B (after 12 years) p-value |
|---|---|---|---|---|---|---|---|---|---|
| Plasma glucose (mg/dl) | FPG | 150.7 ± 1.86 | 154.6 ± 2.21 | 0.213 | 149.5 ± 1.77 | 153.3 ± 3.01 | 0.179 | 0.628 | 0.842 |
| PPG | 191 ± 5.03 | 194.5 ± 5.22 | 0.511 | 189.6 ± 4.82 | 204 ± 10.94 | 0.128 | 0.788 | 0.332 | |
| HbA1c % | 7.86 ± 0.14 | 7.56 ± 0.14 | 0.15 | 7.98 ± 0.15 | 7.71 ± 0.24 | 0.359 | 0.385 | 0.452 | |
| PBMC ROS (geomean of DCF/5 × 105 cells) | 96.31 ± 0.90 | 96.18 ± 0.66 | 0.812 | 98.80 ± 1.69 | 112.6 ± 1.50 | 0.0002 | 0.642 | ˂0.0001 | |
| MDA (nmol/ml) | 2.69 ± 0.03 | 2.63 ± 0.03 | 0.497 | 2.76 ± 0.21 | 3.24 ± 0.08 | 0.038 | 0.769 | ˂0.0001 | |
| AGE (µg/ml) | 2.55 ± 0.07 | 2.65 ± 0.05 | 0.381 | 2.67 ± 0.13 | 3.59 ± 0.25 | 0.002 | 0.791 | ˂0.0001 | |
| NAD+(pmol/106 RBC) | 47.19 ± 1.09 | 47.71 ± 0.86 | 0.322 | 45.29 ± 0.92 | 39.92 ± 0.75 | 0.003 | 0.231 | 0.0007 | |
| NADH (pmol/106 RBC) | 4.42 ± 0.16 | 3.95 ± 0.16 | 0.225 | 4.52 ± 0.10 | 5.56 ± 0.22 | 0.003 | 0.571 | 0.0002 | |
| NAD+/NADH | 10.85 ± 0.48 | 12.72 ± 0.72 | 0.152 | 10.5 ± 0.38 | 7.64 ± 0.66 | ˂0.0001 | 0.133 | ˂0.0001 | |
| 2,3-DPG (µmol/g Hb) | 17.17 ± 0.30 | 17.48 ± 0.54 | 0.615 | 16.20 ± 0.77 | 13.42 ± 0.53 | 0.012 | 0.131 | 0.0002 | |
| VEGF (pg/ml) | 92.33 ± 1.23 | 93.07 ± 2.16 | 0.722 | 94.18 ± 1.37 | 131.7 ± 3.09 | <0.0001 | 0.804 | ˂0.0001 |
Table 4: Biochemical parameters of Group-A and Group-B at the time of diagnosis of T2DM and 12 years after treatment of Group-A (with antihyperglycemic drugs, B-vitamins, ascorbic acid and α-tocopherol) and group-B (only with anti-hyperglycemic drugs).
The Surrogate markers of oxidative stress (ROS and MDA), advanced glycation end product (AGE) formation and vascular endothelial growth factor (VEGF) were found to be further increased towards the development and severity of the disease. However, the ratio of oxidized and reduced co-factors (NAD+/NADH) and red cell 2, 3-DPG activity were found to be decreased with the disease severity.
Comparison of group-A and group-B in the present study showed no statistically significant differences for baseline parameters of surface roughness and osmotic fragility, whereas such parameters were found to be increased in group-B (non-supplemented group) compared to group-A (supplemented group) (Table 5). Study also demonstrated more number of dysmorphic red cells in the blood sample of group-B (Figures 1 and 2).

Figure 1: Scanning electron microscopic appearance of Red Blood Corpuscles (RBCs). (A) Representative image of RBCs from group- A. (B) Representative image of RBCs from group-B. (C) Representative image of RBCs from group-A (after 12 years of treatment with anti-hyperglycemic drugs and vitamins). (D) Representative image of RBCs from group-B (after 12 years of treatment with anti-hyperglycemic drugs only). In case of every images. The size bar corresponds to 2 μm and magnification-5.00 KX.

Figure 2: Kaplan–Meier survival analysis of progression to DR among an individual in group-A, who was treated with antihyperglycemic drugs and vitamins (B-vitamins, ascorbic acid and α-tocopherol) and individuals in group-B who received only antihyperglycemic drugs for 12 years.
| Parameters | Particulars | Group-A (Baseline) N=41 | Group-B (Baseline) N=48 | Group A vs. Group B (Baseline) p-value | Group-A (After 12 years) N=41 | Group-B (After 12 years) N=48 | Group A vs. Group B (after 12 years) p-value | Group-A baseline vs. Group-A (after 12 years) p-value | Group-B baseline vs. Group-B (after 12 years) p-value |
|---|---|---|---|---|---|---|---|---|---|
| Rq | 35.95±1.69 | 37.04 ± 1.70 | 0.655 | 36.90 ± 1.61 | 44.17 ± 1.70 | 0.006 | 0.355 | 0.01 | |
| Ra | 25.42±1.32 | 25.73 ± 1.34 | 0.86 | 26.10 ± 1.32 | 31.22 ± 1.58 | 0.021 | 0.219 | 0.018 | |
| Rsk | 1.34±0.02 | 1.37 ± 0.02 | 0.27 | 1.38 ± 0.04 | 1.46 ± 0.03 | 0.01 | 0.631 | 0.04 | |
| Rku | 1.46±0.05 | 1.48 ± 0.05 | 0.86 | 1.49 ± 0.04 | 1.76 ± 0.11 | 0.038 | 0.213 | 0.021 | |
| Osmotic Fragility | Starting | 5.44±0.09 | 5.37 ± 0.08 | 0.586 | 5.60 ± 0.07 | 6.07 ± 0.07 | 0.0002 | 0.189 | <0.0001 |
| (conc. Of NaCl) | Complete | 4.44±0.11 | 4.41 ± 0.15 | 0.856 | 4.69 ± 0.14 | 5.12 ± 0.14 | 0.047 | 0.243 | 0.003 |
Table 5: Red blood corpuscles surface roughness, osmotic fragility of Group-A and Group-B at the time of diagnosis of T2DM and 12 years after treatment of Group-A (with anti-hyperglycemic drugs, B-vitamins, ascorbic acid and α-tocopherol) and group-B (only with anti-hyperglycemic drugs).
Results demonstrated positive correlations of VEGF with increased ROS,MDA, AGE and negative correlations with decreased NAD+/ NADH and 2,3 DPG at base line of group-A and B. Similar strain was observed in group-B after 12 years, whereas group-A showed no significant correlations.
CMT measurement at base line and 6 years after treatment showed no statistically significant differences. The visual acuity in group-B was found to be decreased after 6 years and 12 years of follow up, whereas group-A demonstrated no significant alteration in VA after 6 and 12 years of follow up (Tables 6-9).
| Parameters | Group-A (Baseline) N=200 | Group-B (Baseline) N=200 | Group A vs. Group B (Baseline) p-value | Group-A (After 12 years) N=187 | Group-B (After 12 years) N=200 | Group A vs. Group B (after 12 years) p-value | Group-A baseline vs. Group-A (after 12 years) p-value | Group-B baseline vs. Group-B (after 12 years) p-value |
|---|---|---|---|---|---|---|---|---|
| CMT (µm) | 244.4 ± 1.56 | 246 ± 1.48 | 0.297 | 250.1 ± 4.41 | 255.6 ± 7.91 | 0.278 | 0.812 | 0.131 |
| VA (log MAR) | 0.224 ± 0.01 | 0.232 ± 0.01 | 0.669 | 0.242 ± 0.02 | 0.342 ± 0.03 | 0.044 | 0.314 | 0.004 |
Table 6: Retinal structural and functional parameters of Group-A and Group-B at the time of diagnosis of T2DM and 12 years after treatment of Group-A (with anti-hyperglycemic drugs, B-vitamins, ascorbic acid and α-tocopherol) and group-B (only with anti-hyperglycemic drugs).
| Parameters | Baseline VEGF level (pg/ml) of Group-A (N=200) | VEGF level (pg/ml) of Group-A after 12 years (N=187) | ||
|---|---|---|---|---|
| Correlation Coefficient (r) | Level of Significance (p) | Correlation Coefficient (r) | Level of Significance (p) | |
| PBMC ROS (geomean of DCF/5 × 105 cells) | 0.66 | <0.0001 | 0.2 | 0.29 |
| MDA (nmol/ml) | 0.62 | 0.001 | 0.18 | 0.33 |
| AGE (µg/ml) | 0.49 | 0.008 | 0.24 | 0.22 |
| NAD+/NADH | -0.59 | 0.004 | -0.24 | 0.24 |
| 2,3-DPG (µmol/g Hb) | -0.52 | 0.006 | -0.26 | 0.19 |
Table 7: Correlation of vascular endothelial growth factor with different biochemical parameters at base line and after 12 years of treatment (with anti-hyperglycemic drugs, B-vitamins, ascorbic acid and α-tocopherol) in Group-A.
| Parameters | Baseline VEGF level (pg/ml) of Group-B (N=200) | VEGF level (pg/ml) of Group-B after 12 years (N=200) | ||
|---|---|---|---|---|
| Correlation Coefficient (r) | Level of Significance (p) | Correlation Coefficient (r) | Level of Significance (p) | |
| PBMC ROS (geomean of DCF/5 × 105 cells) | 0.79 | <0.0001 | 0.67 | <0.0001 |
| MDA (nmol/ml) | 0.47 | 0.008 | 0.7 | <0.0001 |
| AGE (µg/ml) | 0.59 | 0.001 | 0.6 | 0.001 |
| NAD+/NADH | -0.45 | 0.009 | -0.51 | 0.006 |
| 2,3-DPG (µmol/g Hb) | -0.64 | 0.001 | -0.5 | 0.009 |
Table 8: Correlation of vascular endothelial growth factor with different biochemical parameters at base line and after 12 years of treatment (with only anti-hyperglycemic drugs) in Group-B.
| Parameters | Very mild NPDR (N=32) Supplemented group | Mild to Moderate NPDR [N={53(mild NPDR)+ 39 (moderate NPDR)}] Non-supplemented group | Level of significance (p value) |
|---|---|---|---|
| PBMC ROS (geomean of DCF/5 × 105 cells) | 101.7 ± 1.01 | 116.40 ± 2.31 | 0.004 |
| MDA (nmol/ml) | 2.99 ± 0.12 | 3.51 ± 0.14 | 0.034 |
| AGE (µg/ml) | 2.81 ± 0.15 | 3.79 ± 0.26 | 0.021 |
| NAD+/NADH | 9.86 ± 0.39 | 7.02 ± 0.29 | 0.0002 |
| 2,3-DPG (µmol/g Hb) | 15.89 ± 0.76 | 12.12 ± 0.91 | 0.031 |
| VEGF (pg/ml) | 104.18 ±2.42 | 134.9 ± 3.59 | <0.0001 |
Table 9: Biochemical scenario behind the development of the disease.
Thirty-two (17.11%) among 187 subjects of the cohort (Group A) developed very mild microangiopathy, whereas 13 patients lost the follow-up due to migration and allergic reaction to the medication. Among 200 diabetic control subjects (Group B) who did not receive the above-mentioned vitamins supplementation, 92 (46%) patients developed mild-to-moderate (53 subjects with mild NPDR and 39 subjects with moderate NPDR) non proliferative DR within this follow-up period of 12 years. Kaplan-Meier survival analysis demonstrated long term vitamin supplementation more protected diabetic subjects (82.89%) from the development of DR, whereas non supplemented group were less protective (54%) against the development of DR.
India ranks second in the prevalence of diabetes mellitus (DM) and the number of people with DM is expected to increase from current 67 million to 79.4 million by 2030 [18]. With the reported prevalence of diabetic retinopathy in India ranging from 17.6% to 28.2%, the number of diabetics with diabetic retinopathy (DR) will increase to 22.4 million in another two decades [19]. This will result in a heavy burden on the health care system because of high cost of anti-VEGF therapy and inadequacy of infrastructure of laser photocoagulation to fight against DR-induced visual impairment. The crucial factor in the development of DR, that is, biochemical derangements resulting in increase in angiogenic VEGF secretion, if modulated, would have meant the DR-related visual loss could be prevented in our country where infrastructural deficiency and financial constrain create a deep concern.
Although two landmarks studies have demonstrated the beneficial effects of the tight control of hyperglycemia leading to the delay, or even, the prevention of the onset of development of diabetic retinopathy [2,20], patients with good glucose control still develop DR. Further studies suggested that even intensive control of blood sugar (HbA1c to 6.5% or less) did not show palliative effect against the initiation of DR [21,22]. Additionally, a few apparent findings draw our attention towards some patients with poor control of their disease having no DR even over long periods of time, while others have developed DR in a short period of time despite good disease control. However, the fact that improving glycemic control reduces the risk of onset and progression of DR, has evidence that shows that there is no glycemic threshold as reflected in glycosylated hemoglobin that is totally palliative against the risk of retinopathy [23]. Our recent study concluded that differential expression of hyperglycemia-induced biochemical derangements dictate the extent of up regulation of VEGF secretion leading to initiation of DR [24]. In brief, the biochemical anomalies of enormous amount of intracellular glucose in retinal tissues, which are insulin independent for intracellular transport of glucose, flow in various ways, as follows –
• Gradually faster anaerobic glycolysis→altered lactate/pyruvate ratio and NAD/NADH ratio→ reductive stress in cellular level→ deficiency of NAD→inhibition of glycolysis and citric acid cycle;x
• increased generation of lactate→lowering of cellular pH →reduced action glutamate transporter from synaptic space of retinal neurons →glutamate toxicity→increased activation of NMDA receptors→ increased concentration of intracellular Ca++→increased activation of phopholipase enzymes→ increased membrane lipid peroxidation→ increased malondiadehyde (MDA) and advanced lipoxidation end products→ structural and functional alteration of red blood cells and diminished activity of G6PD→reduced function of deformed, rough and rigid red blood cells→localized tissue hypoxia→ increased secretion of VEGF;
• Unutilized huge intracellular glucose → increased nonenzymatic glycation → increased advanced glycation end products (AGEs) formation→AGE + receptor of AGE (RAGE) interaction→activation of NADPH oxidase → increased production of reactive oxygen species(ROS) in pericytes and endothelial cells→oxidativestress→ upregulation of NF-kB→upregulation of VEGF.
Previous studies of ours demonstrated the role of hypoxia-induced anaerobic glycolysis in differential expression of VEGF to be responsible for initiation of DR [25]. Surprisingly, it now becomes clearly obvious that advancement of anti-VEGF therapy, which is being used as a strong weapon to fight against DR-induced vision loss, produces little or no discernible effect in a significant portion of diabetic patients with diabetic macular edema. This finding necessitates the importance of finding other sources of tissue hypoxia. Considering the contradictory results of different experimental studies related to the pathogenesis of DR [26,27], we were led to propose that alterations in structure and function of red blood cell (RBC) produce hypoxia at the retinal capillary beds of diabetic patients and evoke earliest microvascular dysfunction. The red blood cell must be able to squeeze through the tight spots in the microcirculation during its numerous passages around the capillary beds of retina. Under the biochemical background of increased lipid peroxidation, advanced glycation of end product formation and oxidative stress induction, RBCs lose their plasticity, fluidity and oxygen carrying capacity resulting in localized tissue hypoxia at retinal capillary beds.
In this unblended trial, 187 among 400 patients of diagnosed type 2 DM had been treated since their diagnosis and commencement of treatment in 2004, with anti-hyperglycemic medication along with Bvitamins, Vitamin-C and Vitamin-E. Conventional control of hyperglycemia with average glycosylated hemoglobin (HbA1c) between 7 – 7.8% did not experience any significant visual disturbance except age-related correction of refractive errors and did not show any symptom of diabetic retinopathy on dilated fundoscopic examination, digital fundus photography, fluorescein angiography and ocular comprehensive tomography except 32 (17.11%) among this group of patients, languishing for a similar duration of the disease and glycemic status, developed mild microangiopathy characterized by presence of micro aneurysms in the posterior pole of retina. Remarkably, a similarly affected, 200 patients who maintained almost similar glycemic status without intake of vitamins supplementation throughout this yearly follow-up period of 12 years, were seen to have developed mild to moderate NPDR in 92 (46%) patients.
Measurements of biochemical parameters of the postulated detrimental pathways in 200 diabetic subjects who had not received regular vitamins B,C and E, had demonstrated significantly higher levels of lipid peroxidation, advanced glycation end products formation, oxidative stress induction and VEGF secretion, compared to 187 patients who were supplemented with these vitamins. This supplemented group-A did not show positive correlation of VEGF level with ROS, AGEs, MDA, NAD+/NADH and 2,3,DPG after 12 years of treatment with the above-mentioned vitamins. The implicated biochemical mediators are considered to contribute to death of neurovascular cells of retina leading to formation of a cellular capillaries and subsequent tissue hypoxia. Here, the dysfunction of RBCs, the famed cells for their finest ability to squeeze through some tight spots in the microcirculation around the body, could not provide oxygen to the ischemic retina owing to loss of their fluidity and flexibility. Under the systemic environment of non-enzymatic glycation, lipid peroxidation, oxidative stress and inflammation, structural alterations to the lipid bilayer, the actin-spectrin cortex and integral proteins of RBC membrane deform these cells in shape and size and, consequently, these cells become nonfunctional and fragmented. So the RBCs are believed to play an important role for generation of hypoxia during the development of DR.
According to the projections of our recent study, a posteriori to the diagnosis of diabetes mellitus, smooth and uninterrupted running of glycolysis and citric acid cycle, prevention of lipid peroxidation, advanced glycation end product formation and free radical production may ameliorate the micro vascular complications of this disease.
Systemic administration of riboflavin and nicotinamide assure the supply of oxidized cofactors in the body, while pyridoxamine prevents non-enzymatic glycation, and thiamine assists oxidative decarboxylation of pyruvate to acetyl-CoA within mitochondria by pyruvate dehydrogenase enzyme complex; and thiamine also acts as a co-enzyme of transketolase which is necessary to oxidize glucose completely to CO2 via the pentose phosphate pathway. Thus, thiamine diphosphate eliminates the inhibition of pyruvate metabolism that attenuates lethal effects of lactic acidosis. Experimental animal study has shown that benfotiamine, a derivative of thiamine (vitamin B1) blocks the major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy [28]. A recent work on animal models have demonstrated the beneficial effect of prolonged release formulation of niacin (vitamin B3) to prevent apoptosis, expression of vascular cell adhesion molecules-1, VEGF, VEGFR and increased tight junction proteins to lower blood-retinal barrier breakdown in DM [29]. Essential enzymes of glycolysis and citric acid cycle that catalyze group transfer and other reactions require, in addition to their substrate, a second organic molecule known as coenzyme, without which they are inactive. Activity of citric acid cycle is dependent on the supply of oxidized dehydrogenase cofactors, e.g. NAD+. Active niacin is nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+). The nicotinamide nucleotides play a widespread role as coenzyme to many dehydrogenase enzymes acting both in cytosol and mitochondria. Active thiamine, e.g. thiamine diphosphate plays as an essential coenzyme in oxidative decarboxylation of ketogluterate and pyruvate in citric acid cycle, especially, in transketolase reactins in the pentose phosphate pathway. Active riboflavin is flavin mononucleotide (FMN) and flavin adenine dinucleotide, which act as prosthetic group of oxidoreductase enzymes like mitochondrial glycerol-3-phosphate dehydrogenase, in transporting reducing equivalents from cytosol into mitochondria and succinate dehydrogenase in citric acid cycle. Pyridoxal phosphate is active vitamin B6, which by entering into Schiff base combination between its aldehyde group and amino group of an α-amino acid, facilitates changes in the three bonds of α-amino carbon to inhibit non-enzymatic glycation. Pyridoxal 5’-phosphate is also very effective in inhibiting lipid glycation. The previous experimental study demonstrated that AGE-inhibitor pyridoxamine acted as a palliative against diabetes-induced retinal vascular lesions, including thickening of the basement membrane, and acellular capillaries in diabetic rats [30].
So, continuity of glycolysis and citric acid cycles, inhibition of nonenzymatic glycation, prevention of lipid peroxidation and oxidative stress generation, improvement in structural and functional stability of RBCs are some of the curatives that seem to ameliorate the deranged secretion of VEGF and its receptor 2 in supplemented group with thiamine, riboflavin, niacin, pyridoxal phosphate, ascorbic acid and α- tocopherol. The previous studies have advocated the beneficial effects of supplementation of different nutritional components and antioxidants in prevention of progression and development of agerelated macular degeneration and diabetic retinopathy [31-33].
This study, probably a pioneering one, observes the effects of vitamin B, C and E on the surrogate markers of responsible biochemical derangements, and the structural and functional capabilities of RBCs related to development of DR in diabetic subjects for a considerably longer period of time.