Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2012) Volume 5, Issue 12
Probability-based localisation scoring of fragment mass-spectrum phosphorylation site identifications has become common practice to confirm search engine modification assignments, and indicate the degree of certainty with which they are defined. Localisation of modifications other than phosphorylation is also required but is less commonly supported by current tools. These other modifications, such as hydroxylation, may have broad aminoacid specificity, and can be misassigned when the correct specificity is not considered in an MS database search. In addition, localisation software is often specific to a particular MS/MS search engine, and cannot be used to localise modifications identified by multiple search engines. ModLS, a new tool within our freely-available Central Proteomics Facilities Pipeline (CPFP), applies a localisation scoring method to arbitrary post-translational modifications (PTMs). As well as localising PTMs based on amino-acid specificities are included in the initial search, ModLS can automatically consider additional specificities from UniMod. This can help avoid ‘correct modification, incorrect amino-acid’ errors which can occur when data is searched using only a subset of PTM specificities. Localisation scoring can be performed on the results from any search engine incorporated within the pipeline, or where the output of individual search engines is combined to give increased coverage. We demonstrate the performance of ModLS using a publicly available phosphorylated peptide dataset, showing that it outperforms the recently characterised Mascot Delta Score approach for CID and MSA data, and is comparable for HCD data. In addition, we show the utility of automatic specificity expansion using hydroxylated and methylated peptide data. ModLS is a user-friendly localisation tool for arbitrary modifications. Its inclusion within CPFP allows PTM localisation to be performed quickly and easily on large or small result sets, from multiple search engines. Specificity expansion, introduced in ModLS, allows misassignments of modifications due to incomplete consideration of specificities to be identified and minimised.
Keywords: PTM; Phosphorylation; Localization; Algorithm; Pipeline; Open-source
MS: Mass Spectrometry; PTM: Post-Translational Modification; PSM: Peptide to Spectrum Match; CPFP: Central Proteomics Facilities Pipeline; FDR: False Discovery Rate; FLR: False localization Rate
Challenges of post-translational modification localization
The growth in Mass-Spectrometry (MS) studies focused on Post- Translational Modifications (PTMs) has resulted in software designed to streamline the analysis of increasingly large datasets. A critical aspect in PTM studies is the confident localization of modifications within a protein. While MS database search engines can reliably identify modified peptides they may report incorrect localization of PTMs within a peptide. Standard scoring schemes are focused on assessing the confidence of the peptide identification and do not necessarily indicate the specificity of the PTM assignment. The increased use of localization scoring for phosphorylation assignments has been primarily driven by methods published by two groups. Beausoleil et al. [1] presented their AScore method for assessing phosphorylation assignment ambiguity and provided software to post-process SEQUEST results. In the related PTM Score approach described by [2], Olsen et al. [3] has subsequently become prominent due to its incorporation in the SILAC quantitation tool MaxQuant [4]. These methods, which are based on binomial probability calculations, along with others such as SloMo [5] and PhosphoRS [6] perform localization in a post-search re-analysis of Peptide to Spectrum Matches (PSMs). Alternative methods make use of search-engine scores to calculate localization ambiguity. The Mascot Delta Score, which uses the native scoring function of the Mascot search engine to calculate the confidence of PTM localization, is an example of this method [7]. Recently, commercial software such as Scaffold PTM (Proteome Software) has begun to incorporate these methods. Each tool has strengths and weaknesses, related to the localization scoring method applied, and the implementation of the software itself [8].
Key problems with many existing modification localization tools are that they are either restricted to specific PTMs (commonly phosphorylation), or can only analyse the output from a single database search engine. Users are restricted to certain localization methods depending on their choice of search software, and/or can only localize phosphorylation sites. Recent data analysis pipelines, including our Central Proteomics Facilities Pipeline CPFP [9], can now automatically search a dataset with multiple search algorithms and combine their results. This process can result in increased peptide and protein identification rates but existing PTM localization tools cannot be applied to the data easily. Through its integration into CPFP, our tool ModLS supports localization of PTM results from multiple search engines, and can operate on meta-search results which combine output from these search engines. ModLS can perform localization scoring for any PTM selected as a variable modification in a search, from those defined in the comprehensive UniMod database [10] which is imported by CPFP.
Ease-of-use and the ability to manually review localization options are other important considerations in the selection of a tool. Simple scripts such as PhosCalc score localizations, but do not provide a simple way to comprehensively review the different assignments considered, with annotated spectra etc. The AScore web server annotates localization neatly, but is difficult to use. Database search must be performed separately and the results uploaded to the tool in a restrictive format.
Automatic specificity expansion for PTMs
A novel extension to ModLS, differentiating it from existing tools, is that of automatic specificity expansion. Existing software considers only localizations involving specific hard-coded PTMs (such as phosphorylation of Ser/Thr/Tyr) or the PTM specificities chosen as variable modifications during the search. This is generally sufficient for phosphorylation studies which are routinely focused on presence of the phosphate group on Ser/Thr/Tyr. Phosphorylation of other amino acids is uncommon and rarely considered.
Isobaric oxidation and hydroxylation, now recognised as important PTMs in signalling processes [11], can occur on many amino acids either in-vivo or as an artefact during sample preparation [12]. UniMod currently lists 13 out of the 20 amino acids as having the potential to be oxidised post-translationally or as an artefact of sample processing. For wide-ranging studies on oxidative modifications, this presents significant challenges. If all oxidation possibilities are used as variable modifications then the search-space for MS/MS spectrum matching increases dramatically, affecting run-times and specificity. It is not usually practical to perform unrestricted PTM searches for large datasets.
A common strategy in this situation is to perform one or more searches, each with a limited set of variable modifications. In many cases, these searches may only cover PTM specificities known to be commonly observed or interesting in the context of the experiment. For example, a user may search a dataset only for proline hydroxylation, common on collagen. In this case, a PTM localization may be assigned incorrectly. Consider a tryptophan residue oxidised during a gel-digest procedure, proximal to a proline. The fragment spectrum may support an incorrect indication of proline hydroxylation.
Multiple searches can be performed, each with a different subset of specificities, and different localizations may be suggested for the same spectrum in different searches. This allows the situation previously to be resolved, by inspecting conflicting assignments for the same spectrum. The true localization will likely result in a higher assignment score. This procedure is lengthy and prone to error where users do not identify and resolve conflicting assignments for a spectrum. Mono/Di/Tri-Methylation can offer similar challenges. In studies examining arginine methylation, it is important to consider the possibility of incorrect assignment of lysine methylation [13].
ModLS introduces automatic specificity expansion which can help highlight these errors. A search is performed with a limited set of variable modifications, but all possible specificities are considered at the localization scoring step. This allows incorrect assignments that are due to incomplete consideration of PTM specificity to be resolved. The procedure does not replace nonspecific PTM searches, since it only operates on identifications from the initial specificity-restricted search. It cannot identify additional spectra. However, we propose that the procedure is a valuable aid for users studying PTMs such as oxidation, on large datasets that are not feasible to analyse with nonspecific search tools.
ModLS implements a variant of the PTM Score algorithm described by Olsen et al. [3]. A dataset of one or more MS/MS peak list or MzXML files is uploaded to CPFP. Peptide identifications from multiple search engines (currently Mascot, X! Tandem and OMSSA) are validated and combined using tools from the ISB Trans-Proteomic Pipeline (TPP) [14]. Results are processed for False Discovery Rate (FDR) assignment using target-decoy sequence databases and q-value filtering [15], before being imported into a MySQL database. The user performs ModLS PTM scoring by submitting a task against the search results via the CPFP web interface. This creates a job executed by CPFP on the server, or a local or remote compute cluster (depending on the configuration of CPFP).
For each peptide to spectrum match, above a specified FDR, with at least one PTM assignment ModLS calculates all combinations of localization of the PTMs present on the peptide. Standard localization uses only the PTM specificities chosen as variable modifications in the search to generate these combinations. If automatic specificity expansion is enabled, the combinations will be increased to include all possible specificities defined in UniMod, for the PTMs chosen in the search.
For each localization combination, a PTM Score is calculated matching the theoretical spectrum to the experimental spectrum. The theoretical ions considered are then-terminal and c-terminal fragmentation series, losses of NH3 and H2O, plus informative neutral losses from the UniMod PTM definition (e.g. H3PO4 for phosphorylation). Neutral losses corresponding to the entire mass of the PTM are excluded as they generally do not help determine localization. A theoretical ion that is modified, and loses the entire mass of the modification, may erroneously match to a spectrum peak generated by an unmodified sequence ion of the correct localization. For CID and HCD instruments b and y series ions are considered. When viewing results in the CPFP web interface additional MH, immonium and internal fragments may be annotated, but these are not used by to calculate the PTM score.
ModLS implements the PTM Score method of Olsen et al. [3] except that the peak depth is varied from 1 to 10 peaks per 100 m/z window and the maximum resulting value is taken as the final PTM Score. The PTM Score calculation is:
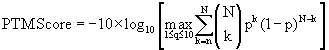
where n is the number of matched theoretical ions for peak depth q peaks per 100th window, N is the total number of theoretical ions, and p is the probability of a match at random within a 100 m/z window:
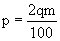
where m is the symmetric MS/MS mass tolerance and q is the peak depth under consideration.
All localizations are ranked by PTM Score before calculating the final Modification localization Score, which is a measure of localization ambiguity analogous to the AScore. For the top-ranked localization the ModLS value is the difference in PTM Score between this and the second ranked. A ModLS value of 0 indicates that no spectral evidence was found that uniquely localizes the PTM(s). For lower rank combinations, the score is the difference between that combination’s PTM Score and the highest PTM Score, indicating how much ‘worse’ the assignment is than the top scoring hit. When all localizations have been scored, a probability is also derived for each individual possible PTM site, as detailed in [3].
ModLS is written in Perl, as a module of the CPFP analysis pipeline. It can perform localization scoring of multiple PSMs in parallel on multi-core machines. Input spectra and PSMs are retrieved from the CPFP MySQL database. Results from ModLS are stored in the database and displayed in CPFP’s peptide results viewer, which allows PSMs to be easily filtered and exported to Excel. Any disagreement on the most likely PTM localization between the search engine results and ModLS is clearly highlighted. Detailed ModLS results can be accessed for each PSM. The top 10 localizations per peptide are presented with annotated spectra for manual review. Users can navigate to a detailed spectrum viewer that presents all fragment ion matches in tabular format, as well as a larger more detailed annotated spectrum figure. For publication users may access a web page displaying annotated spectra for multiple selected peptides. This view may be easily saved as a PDF file from modern browsers, allowing spectra to be supplied as supplementary material for manuscript submissions.
Performance for phosphorylated peptide localization
To demonstrate the strength of applying CPFP and ModLS to phosphorylation studies, we analysed a publicly available gold-standard dataset of known synthetic peptides. We compared the performance of our tool to the Mascot Delta Score (MD Score) method, which was demonstrated to outperform the A-Score algorithm by Taus et al. [6] and Savitski et al. [7]. MDScore resulted in a higher number of phospho-peptide matches for a given FLR than A-Score in these studies. Like A-Score and various PTM Score implementations, the method is search-engine specific. MD Score can only be used to localize Mascot search results, while ModLS localizes results from Mascot, X! Tandem and OMSSA combined within CPFP. However, MD Score is now tightly integrated into a database search engine – the commercial Mascot Server version 2.4 includes MD Score PTM localization by default. This ensures that MD Score is amongst the most widely-used localization methods, and is appropriate to compare to the tightly integrated ModLS/CPFP solution since introduction MD Score has been used in various phosphorylation studies e.g., [16,17].
The Savitski et al. [7] dataset consists of 180 synthetic phosphorylated peptides with known phosphorylation sites, combined into 5 mixtures for analysis. During development of ModLS, this was the most comprehensive public gold-standard phospho-peptide dataset available. Its size allows more accurate calculation of false localization rates than smaller peptide mixtures. The same dataset has been used in other studies [18,19]. We analysed data files made publicly available by the authors, covering three fragmentation methods [5]. Collision Induced Dissociation (CID) and Multi-Stage Activation CID (MSA) were acquired on an LTQ-Orbitrap using the Orbitrap detector for MS scans and ion-trap for MS/MS scans. The HCD data were acquired using the Orbitrap for both MS and MS/MS scans. For MSA data, where the instrument detects a dominant neutral loss ion, resulting from loss of the phosphate group from the peptide, additional fragmentation is performed to generate more complete fragment ion series.
Unfiltered MGF files provided by the authors were uploaded to CPFP. Minimal peak picking is performed on import to CPFP, limited to removal of peaks with an absolute intensity<5 units. Further peak-picking is performed during PTM localization only; the ModLS PTM score calculations automatically consider peak depths from 1 to 10 peaks per 100 Da, so limiting the dataset to 6 peaks per 100 Da as in the filtered peak lists [7] would be inappropriate. For the HCD data, we additionally uploaded charged-reduced and de-isotoped data (HCD Reduced), since MD Score performance demonstrated a dependency on processing HCD data in this manner.
Analysis was performed on CPFP version 2.1.03 with searches using Mascot (version 2.3.01, Matrix Science, London, UK), X! Tandem with the k-score plug-in (version 2008.12.01.1) [20], and OMSSA (version 2.1.8) [21]. The UniProtKB human whole proteome sequence database (release 2012_07) [22] was used, as the IPI database from the MD score study is deprecated (http://www.ebi.ac.uk/IPI). The UniProt database provides full-coverage of the synthetic peptides, which were generated from human protein sequences. Reversed decoy sequences were appended for False Discovery Rate (FDR) estimation. Trypsin was the selected digestion enzyme and up to 3 missed cleavages were allowed. Carbamidomethyl cysteine was specified as a fixed modification, while oxidised methionine, protein n-terminal acetylation, and phosphorylated serine/threonine/tyrosine were chosen as variable modifications. Precursor mass tolerances were 20 ppm in all cases, while fragment tolerance was 0.02 Da for the HCD dataset, and 0.5 Da for all other data. Isotope selection errors of +1 and +2 Da were considered during the searches. Note that search parameters are not identical to Savitski et al. [7] as we used a combination of search engines, with different optimal settings than Mascot alone.
Post-processing of results in CPFP was with default parameters, excepting that PeptideProphet was instructed not to penalize Mascot results with high homology between top and second ranked identifications. ModLS was run against meta-search results, where output from each search engine is combined using iProphet from the TPP. For each dataset, the combined PSMs were filtered to a 1% FDR for spectrum identification using decoy hits. Phosphorylated peptide identifications and localizations were matched to the reference list from the original Savitski et al. [7] study. False Localization Rates (FLRs) amongst these phosphorylated peptides were then calculated for varying ModLS score thresholds, and are compared to the results from Savitski et al. [7]. PSM results are provided in supplementary tables S1-S6. PDFs containing annotated spectra for all identified phosphorylated peptides are available on our website (http://cpfp.sourceforge.net/modls).
Table 1 lists the number of correctly localized spectra, and unique phosphorylated peptides at a 1% FLR for each fragmentation method. The MD Score values are taken from Mascot Delta Score [7] as the highest correctly localized clustered peptide and spectrum counts for each fragmentation method, from either filtered or unfiltered peak lists. On the CID and MSA data ModLS strongly outperforms MD Score, identifying 32% and 13% more phospho-peptides respectively. An even greater improvement is observed in the number of correctly localized spectra. We note that multi-stage activation increases the number of phosphorylated peptides identified and correctly localized by 10%, from 79 to 87, highlighting the importance of further fragmenting dominant precursor neutral loss ions for labile modifications.
| ModLS | MD Score [5] | Improvement vs MD Score | |||||
|---|---|---|---|---|---|---|---|
| Fragmentation | ModLS Threshold | Peptides | PSMs | Peptides | PSMs | Peptides | PSMs |
| CID | 12 | 79 | 1071 | 60 | 503 | +32% | +121% |
| MSA | 10 | 87 | 1006 | 77 | 535 | +13% | +88% |
| HCD | 27 | 118 | 693 | 85 | 393 | +39% | +76% |
| HCD (Reduced) | 25 | 124 | 740 | 131 | 789 | -5% | -6% |
Table 1: Performance of ModLS for phosphorylation site localization compared to the MD Score method. Peptides and PSMs columns list counts of correctly localized phosphorylated peptides and PSMs for each fragmentation methods. Filtering has been performed to a 1% False Localization Rate (FLR). MD Score values listed are the highest reported by Savitski et al. [3] for filtered or unfiltered data. Results demonstrate that ModLS outperforms MD Score for CID and MSA datasets, and is comparable for HCD data.
HCD performance for ModLS and MD Score are broadly comparable. On the charge-reduced, de-isotoped data ModLS achieves approximately 5% lower performance than MD Score; On unfiltered data, the method greatly outperforms MD Score. ModLS is relatively insensitive to charge-reduction and de-isotoping for HCD data, localizing only 5% more peptides and 7% more spectra vs unfiltered data. The total number of spectra identified by the three search engines in CPFP increased from 1318 to 1401 at a 1% FDR after peak processing, a similar small 6% improvement. This demonstrates that the combination of search engines used by CPFP is less sensitive to peak list filtering for high-mass accuracy HCD data than Mascot alone. HCD fragmentation is clearly preferable to CID methods, identifying additional correctly localized phosphorylated peptides, despite fewer spectra being collected.
ETD search capability is available in CPFP after recent updates, but ModLS has not been optimised or well tested on ETD data. Initial experiments using c and z+1series ions for PTM Score calculation yielded poor results. Correctly identified and localized phosphorylated peptide counts were approximately 40% lower than for the MD Score method. Optimisation of ModLS for ETD datasets will be performed for a future release of CPFP, with reference to related tools such as SlowMo [5], which demonstrates good performance on ETD data using binomial probability based algorithms. Specifically, the authors of SlowMo implement removal the common and abundant charged-reduced product ions from ETD spectra, which complicate ETD spectra significantly. In PhosphoRS, ETD localization was investigated by including or excluding various ion series, with the result that localization based on solely singly-charged c and z-series ions was optimal [6].
Figure 1 shows the relationships between ModLS threshold, the number of correctly localized PSMs and FLR. For CID and MSA, the 1% FLR is achieved by filtering results with a ModLS cut-off of 12 and 10 respectively. This shows that ModLS values are relatively stable across the two fragmentation methods where the same mass tolerance is used. The HCD datasets exhibit a stationary area of approximately 4-5% FDR for ModLS thresholds of 8 to 18. We believe that this behaviour of the FLR vs ModLS relationship for HCD may be due to the assumption in the PTM Score algorithm that fragment ion matches are equally likely to occur across the entire mass range. Others have proposed that this assumption becomes increasingly invalid as lower mass tolerances are used [8]. Nevertheless, the use of the synthetic dataset allows the identification of a suitable ModLS cut-off for a 1% FLR. The number of correctly localized PSMs decrease fairly linearly as the ModLS cut-off is increased. It is clear that use of a ModLS threshold>15 for non-HCD datasets will result in significantly decreased PSM counts without a useful decrease in FLR.

Figure 1: ModLS score versus false localization rate and number of correctly localized PSMs. A) Number of correctly localized PSMs for varying false localization rates and B) the false localization rates for varying ModLS score thresholds on the phosphorylation datasets. As phosphorylated PSMs are filtered with an increasing ModLS score threshold, the false localization rate declines rapidly, while the number of correctly localized PSMs decline slowly. A ModLS score threshold can be selected to achieve a desired false localization rate, or trade-off between FLR and phospho-peptide assignments.
We find that CPFP with ModLS strongly outperforms the MD Score method for localization of phosphorylation on CID and MSA data. The tool is broadly comparable to MD Score for HCD datasets. We aim to provide ETD capability in a future release.
Utility of automatic specificity expansion
Having established the performance of ModLS scoring for phospho-site localization, we now present examples demonstrating the utility of automatic specificity expansion. Oxidation, now recognised as an important PTM in signalling processes, as well as an indicator of stress and aging of proteins, can occur on many amino acids either in-vivo or artefactually during sample preparation. The UniMod database currently lists 13 out of the 20 amino acids as susceptible to oxidations either post-translationally or artefactually. As previously detailed by Cockman et al. [23], the enzyme FIH hydroxylates asaparagine residues within the C-terminal activation domain of Hypoxia Inducible Factor 1-alpha (HIF1α) as well as asaparaginyl residues within the Ankyrin Repeat Domain (ARD) of many ARD containing proteins. Recently hydroxylation of histidine [24] and aspartate [25] residues within ARDs has been identified.
As stated previously, searching with very wide PTM specificity increases the search-space for MS/MS spectra dramatically, adversely affecting search speeds and decreasing specificity. To avoid these problems, we can perform multiple searches using restricted specificities. Specificity expansion is then used to confirm the specificities of reported hydroxylations, resolving conflicts between assignments in the restricted individual searches.
We analysed a dataset containing hydroxylated Rabankyrin (ANKFY1) protein, a substrate of FIH with known hydroxylation sites. A search was performed specifying oxidation of Met, His, and Trp variable modifications. These specificities are most commonly associated with oxidation artefacts from protein digestion and sample storage. However, ANKFY1 is known to be enzymatically hydroxylated at various Asn residues. In figure 2, we show the various localizations considered by ModLS for a single spectrum that was assigned to the peptide SALFLLEHQAADINVR by the CPFP database search engines and contained a single hydroxylation/oxidation. The ModLS output shows that the database search-engine assignment was for His hydroxylation. Alternative localizations were considered and scored by ModLS specificity expansion with the most unlikely assignments excluded from display. The most probable localization, with the highest ModLS and PTM Scores, is on Asn-649 which is the correct known site of FIH mediated hydroxylation. The ModLS localization score of 10 corresponds to an approximately 1.1% FLR from the phospho-peptide CID benchmark. By examining the images of spectra, we clearly see that the incorrect His assignment by the database search engine is missing matches for the y8 and y9 ions. Without specificity expansion, we could have reported an incorrect localization of the hydroxylation. The web interface of ModLS allows users to access far larger detailed spectral images, with ion-match tables to manually review each possible localization. Within the same dataset, we were also able to assign hydroxylation to Asn-798 with a ModLS score of 50, without the initial search having considered the correct specificity.

Figure 2: Automatic specificity expansion results in accurate localization of hydroxylation. ModLS localization with specificity expansion of known asparagine hydroxylation in the Asn-649 peptide of Rabankyrin (ANKFY1). This figure presents the various possible localizations considered by ModLS for hydroxylation on a single peptide to spectrum match. The original database search was conducted without specifying Asn hydroxylation as a variable modification and the search engine incorrectly assigned His oxidation (bottom row). Using specificity expansion ModLS correctly identifies the Asn-649 hydroxylation as the most likely, highest scoring, assignment (top row). This demonstrates how specificity expansion can identify and prevent reporting of PTM assignments to an incorrect amino acid.
To further demonstrate the benefits of specificity expansion on a different PTM, we examined spectra from an arginine methylation study [13] with ModLS. This study was focused on the identification of arginine mono and di-methylation sites, but methylation occurs at additional specificities, including lysine. Where a search is performed using mono/di-methyl arginine as variable modifications true lysine methylation may be assigned incorrectly to arginine. Since the study used heavy methyl-SILAC labelling, a search for Arg mono/di-methylation requires 6 variable PTMs with the inclusion of light and heavy methionine oxidation. Addition of the lysine variable modifications would require a total of 10 variable modifications. Since the addition of variable modifications has a multiplicative effect on search complexity and duration such a large number of variable PTMs may not be practical with large datasets.
We took a single data file from the Orbitrap analysis of HILIC separations in the Uhlmann et al. [13] study which is available online in the PeptideAtlas public repository. This file was analysed using CPFP specifying light and heavy mono and di-methyl arginine as variable PTMs, plus light/heavy methionine oxidation. Lysine specifity for methylation was not included. Table 2 summarises three cases where ModLS specificity expansion was employed to correctly localize lysine methylation, which had been misassigned to arginine in the initial search. Since methylation typically interrupts tryptic cleavage these localizations are supported by additional information other than the improved fragment-ion matching considered by ModLS. Within the original study, methyl-SILAC labelling was used to confirm the presence of methylation on the peptide, adding to the confidence of our assignments. Supplementary figures S1-S3 provide localization detail for these cases.
| Peptide | PTM | Search Localization | ModLS Localization | ModLS Score | Corroborating Evidence | Supp. Figure |
|---|---|---|---|---|---|---|
| DSRPSQAAGDNQ GDEAKEQTFSGGTSQDTK | Dimethyl | R3 | K17 | 45 | Many b/y ions | S1 |
| MDSTEPPYSQKR | Dimethyl | R11 | K10 | 18 | b-11 ion Miscleavage | S2 |
| QSGYGGQTKPIFR | Methyl | R13 | K9 | 14 | Many b/y ions Miscleavage | S3 |
Table 2: Examples of ModLS localization with automatic specificity expansion on the Uhlmann et al. [13] methylation dataset. Initial search was performed for Arg methylations only. ModLS corrects the localization of the PTM for 3 example peptides. All peptides have corroborating evidence by manual inspection of spectra and/or location of missed tryptic cleavage. Full localization detail is given in the indicated supplementary figures S1-S3.
The above analysis of the dataset including ModLS specificity expansion required 5 min 31 sec to complete, versus 7 min 49 sec for a search that also included lysine specificity. The difference in duration is small for a single data file, but in a large proteome-wide study that may contain in excess of 100 MS runs a large time-saving can be achieved. Note also that the addition of the lysine modifications to the search decreased overall sensitivity, reducing the number of peptide-spectrum matches at a 1%FDR from 1811 to 1760. A non-specific ‘error tolerant’ search using Mascot Server version 2.4, which considers all PTMs in the UniMod database, required 11m 32s. This error tolerant analysis also requires far more manual review of results to exclude spurious matches to implausible PTMs.
We believe that assignment of PTMs to incorrect amino acids, due to incomplete search specificities, is of genuine concern in large PTM studies and not usually considered when applying localization tools. We find that the automatic specificity expansion functionality in ModLS simplifies the discovery of these errors, providing a means to improve the quality of PTM results in large studies. The technique is not yet ready for high-throughput automated analyses without manual review, because the effects of specificity expansion on false discovery and localization rates must studied using a gold-standard dataset. To date a dataset of synthetic modified peptides where the PTM has variable amino acid specificities, such as oxidation or methylation, or similar is not available. We recommend that specificity expansion is used to inform manual review of datasets and raise awareness of the possibility of incorrect assignment of PTMs to the wrong amino acid. The method supplies an alternative to lengthy unrestricted modification searches (such as Mascot error tolerant search), the results of which themselves require extensive manual review for false positives. To span the range of specificities for PTMs of interest, users can perform multiple searches, each with a restricted set of variable modifications. ModLS specificity expansion results can then be used to resolve localization conflicts between these searches.
We have presented and demonstrated our software, ModLS, for the localization of post-translational modifications. ModLS is a novel method as it permits consideration of PTM specificities other than those considered in the original database search of a dataset. We have demonstrated that this can help to reduce errors where a correct PTM mass shift is assigned to an incorrect amino acid. Additionally we have shown that ModLS outperforms the existing MD-Score method for PTM localization on a CID dataset of known phospho-peptides, and is comparable for HCD data.
ModLS is integrated in our Central Proteomics Facilities Pipeline, allowing it to be applied to the results of multiple database search engines. The software is open-source and available via http://cpfp.sourceforge.net A demonstration server is also available at this address.
We acknowledge the Computational Biology Research Group, Medical Sciences Division, University of Oxford for use of their services in this project. We thank Dr. H Mirzaei, UT Southwestern Medical Center for support during this project. DCT and RS were funded by ERC FP7 grant (project 233240) to PJR. DCT was also part-funded by Cancer Prevention Research Institute of Texas award RP120613 to H. Mirzaei, UT Southwestern Medical Center at Dallas. We acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper.