Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2023)Volume 12, Issue 3
In-silico manner has completed a position of amazing significance in the drug discovery area. Molecular docking is a computational method widely used for unexpectedly predicting the binding modes and affinities of small molecules against their goal molecules (normally proteins). Molecular docking has been significantly facilitated by way of dramatic growth in computer power and the growing availability of small molecule and protein databases. Webina is a JavaScript/web assembly library that runs Autodock Vina, a popular software for molecular docking, completely in an internet browser. The docking calculations take area at the user's very own computer as opposed to a remote server. Webina web app includes a handy interface so users can without difficulty setup their docking runs and analyze the effects. A running model of the app may be accessed free of fee. A general 18 synthesized peptide derivatives have been completed for molecular docking studies with 4FGW protein through the use of webina net server. The A2, A3, B1, B2 and B3 had been displaying good activity.
WEBINA 1.0.3; Molecular docking; 4FGW protein; Tri-peptides; In-silico
Molecular docking is a computational system broadly used for rapidly predicting the binding modes and affinities of small molecules in opposition to their target molecules (normally proteins). This in-silico system has accomplished a function of great significance with inside the drug discovery field. Molecular docking has emerged over the last many years and is now taken into consideration as necessary device for CADD and with inside the structural biology field, and has been proven to be extra green than conventional drug discovery methods [1]. Molecular docking has been substantially facilitated via way of means of dramatic increase in laptop strength and the growing availability of small molecule and protein databases.
Many molecular docking programs such as Autodock, Dock, Flexx, Glide, Gold, Surfflex, ICM and Ligandfit have been developed in recent years and successfully used in many drug discovery computer science projects. Coupling consists of identifying ligands that bind within more favorably receptor binding sites and determine their energetically most favorable binding orientations (poses). The term "binding position" is the orientation/confirmation of a ligand relative to its receptor. A union position refers to the conformation of a ligand molecule within the binding site of its target protein that has been experimentally confirmed or a computationally modeled hypothetical conformation. The search algorithm and the scoring function are two important components in determining protein-ligand interactions and identifies the most favorable receptor/ligand binding modes. An ideal search algorithm should be fast and efficient, and the scoring function should be able to determine the physicochemical properties of the molecules and the thermodynamics of the interactions [2].
Webina is a JavaScript/web assembly library that runs AutoDock Vina, a popular molecular docking program, entirely in a web browser. Coupling calculations are performed on the user's own computer rather than on a remote server. To encourage usage, we have embedded the Webina library into our own webina web application. The application includes a convenient user interface that allows users to easily set up their docking runs and analyze the results. A working version of the application.
Description of use
PDBQT receptor/ligand input files: As with the Vina command line, Webina accepts receptor and ligand input files in PDBQT format. The latest version of the Webina app is optional interacts with the PDBQT convert application (included in the Git repository) to convert these files from other formats (e.g. PDB) to PDBQT. However, some advanced users may wish to provide their own PDBQT files. These users include: Users who want finer control over what they type. For example, users who want protonation states, ring conformation shapes, etc. Users who want to access the Webina JavaScript library independently of our easy to use Webina app. The library itself is just Vina compiled to web assembly. It cannot convert files because Vina cannot convert files, so users must provide their own PDBQT files [3].
Receptor PDBQT file preparation: The following steps for those who want to provide their own receptor PDBQT.
• Download the PDB file from the files protein data bank.
• Remove ligands, ions, cofactors, water molecules, etc. which might interfere editing the PDB file in a text editor with docking. (e.g. notepad++) and removing the corresponding ATOM and HETATM records is perhaps the easiest way remove these components.
• Assign hydrogen atoms to the protein model. We recommend using the PDB2PQR server, an online website that adds hydrogen atoms based on custom pH values.
Convert to the PDBQT format: The PDB2PQR server will produce a PQR report that consists of hydrogen atoms. The most effective manner to transform this report to PDBQT is with the loose application Open Babel. Here is an instance command: Obabel-xr-ipqr my receptor. Pqr-o my receptor. PDBQT.
Preparing the ligand PDBQT file: We advocate the subsequent steps for people who want to offer their own ligand PDBQT documents: Obtain a replica of your ligand report in SMILES-string or SDF format. Many molecular databases offer small-molecule documents in those not unusual place codecs. If necessary, you may convert to those codecs the usage of open babel. Many online molecular editors additionally generate SMILES strings through letting customers draw their molecules. Generate 3-D fashions of your ligand. We advocate the usage of this system gypsum-DL to generate awesome fashions that account for exchange ionization, tautomeric, chiral, cis/trans isomeric, and ring-conformational states. Convert to PDBQT. Gypsum-DL will output ligand fashions with inside the SDF format. You can convert those to PDBQT the usage of open babel like this: Obabel-isdf gypsum_output. Sdf-o gypsum_output. PDBQT. It is likewise viable to transform immediately from a SMILES or SDF report the usage of only Open Babel, aleven though Open Babel lacks a number of the functions gypsum-DL provides. Here is an instance command line: Obabel--gen3d-p-ismi ligand. Smi-o ligand. PDBQT [4].
4FGW (Glycerol-3-Phosphate Dehydrogenase, GPD1): There are three distinct objectives for glucose inhibition. They are:
• Glycerol-3-phosphate dehydrogenase (NAD(+)), cytoplasmic.
• 5'-AMP-activated protein kinase subunit beta-1.
• Electro transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial.
Glycerol-3-phosphate dehydrogenase (NAD(+)), cytoplasmic: If mGPD features predominantly with inside the glycerophosphate trip (crimson box), inhibition through metformin might be anticipated to sluggish the elimination of NADH, main to an boom with inside the cytosolic NADH/NAD+ ratio that feeds lower back on Lactate Dehydrogenase (LDH). If flux from glycerol to glucose is significant (blue box), inhibition of mGPD through metformin may also cause accumulation of glycerolthree-phosphate (G-three-P) such that oxidation to Dihydroxyacetone Phosphate (DHAP) through cGPD will become favorable. Whereas mGPD catalyzes this response through donating electrons immediately to the electron shipping chain, cGPD might concomitantly produce NADH, growing the cytosolic NADH/NAD+ ratio, which might feed lower back on LDH. Note that the glycerophosphate trip catalyzes the internet switch of electrons from NADH to ubiquinone (Q) with inside the electron shipping chain with regeneration of the intermediate Dihydroxyacetone Phosphate (DHAP) and G-three-P pools [5]. Reverse flux via cGPD might now no longer be anticipated with inside the absence of an outside supply of G-three-P or oxidation of the cytosolic NADH pool (Figures 1 and 2).

Figure 1: PDB structure of 4FGW.
Structure of Glycerol-3-Phosphate Dehydrogenase, (GPD1): From Sacharomyces cerevisiae.
Classification: Oxidoreductase
Organism(s): Saccharomyces cerevisiae S288C
Expression system: Escherichia coli
Mutation(s): No
Deposited: 2012-06-05
Released: 2012-11-07
Deposition author(s): Aparicio, D., Munmun, N., Carpena, X., Fita, I., Loewen, P.

Figure 2: Experimental data snapshot.
Method: X-ray diffraction
Resolution: 2.45Å
R-value free: 0.207
R-value work: 0.200
R-value observed: 0.200
Software used: WEBINA 1.0.3
Input parameters tab
On first journeying the Webina internet app, the user encounters the "input parameters" tab. This tab includes several subsections that are useful for putting in a Webina run. Input PDBQT files. The "input (PDBQT) files" subsection permits the user to pick their receptor and ligand documents. The user also can optionally specify a known-pose PDB or PDBQT ligand document. This document includes the ligand in its experimentally determined, accurate sure pose (e.g., in line with X-ray crystallography or NMR) [6]. The recognized-pose document plays no role inside the docking calculation; rather, it serves as a fantastic-control reference for comparing Webina-anticipated ligand poses. In our experience, it is often beneficial to first benchmark Webina (or Vina) towards a known ligand earlier than the usage of this system to assess compounds with unknown poses and binding affinities. The "input (PDBQT) files" subsection additionally consists of several alternatives to simplify the process of getting ready/checking out protein/ligand enter documents. The receptor/ligand input files which aren't in the required PDBQT layout, the Webina internet app will optionally try to convert them to PDBQT using the PDBQT Convert app. Interactions among the Webina and PDBQT Convert apps occur at "arm's length" through an iframe. If the receptor documents encompass non-protein residues that could interfere with docking (e.g., a co-crystallized ligand), they could cast off all non-protein atoms. If users do now not have a ligand file, they are able to use an internet-based totally 2D molecular editor to draw their ligand by using hand. PDBQT convert then converts that 2D ligand illustration to a 3-D PDBQT report for docking (Figure 3).

Figure 3: Giving input in Webina.
Docking box
The "docking box" subsection allows customers to specify the vicinity of the receptor wherein Webina must attempt to pose the ligand. This field-shaped volume is normally centered on a recognized protein pocket or cavity in which a small molecule ligand may fairly bind. If the field is huge sufficient to encompass the complete protein, Webina will strive full-floor docking. To simplify the technique of selecting a docking box, the Webina net app routinely shows 3D fashions of the consumer-particular receptor and ligand using the 3Dmol.Js JavaScript library. By default, the receptor and ligand are displayed using caricature and sticks representations, respectively. The person can toggle a surface illustration as required to pick out candidate receptor wallet. A transparent yellow field is superimposed at the structures to suggest the docking-box vicinity. When the person clicks the atoms of the receptor model, the Webina internet app recenters the docking box on the chosen atom. Users can also alter the area and dimensions of the box the usage of textual content fields beneath the molecular visualization (Figure 4) [7].

Figure 4: Docking box representation in Webina.
Other critical parameters
The "other critical parameters" subsection lets in the user to specify the wide variety of CPUs and the exhaustiveness putting. We chose to set those two parameters apart because they are in particular essential in a browser-primarily based setting [8-10]. Users expect command line equipment to consume sizeable computer resources, but they do no longer count on web apps to achieve this. By default, Vina makes use of all to be had CPUs and an exhaustiveness putting of 8. Webina has the same capability to consume CPUs and memory, but many users will desire to adjust those parameters to keep away from impacting the performance of other packages and browser tabs.
Advanced parameters
The "advanced parameters" subsection lets in users to specify many extra parameters that are additionally available through commandline Vina. In our revel in, it's far rarely essential to regulate those parameters, so they're hidden via default.
Run Vina from command line
The "run Vina from command line" subsection goals to assist Vina customers who want to apply the Webina internet app to setup their docking bins and consumer parameters. A text subject provides a ridicule instance of a way to use command line Vina with the particular parameters. Users can replica this case, modify it as wished, and paste it into their command-line terminals to run the preferred calculation with the standard Vina executable. This subsection additionally includes hyperlinks that permit the user to download the receptor/ligand PDBQT documents for command line use.
Starting the Webina calculation
Once customers click on the "start Webina" button, the Webina app will transfer to the "running webina" tab whilst webina executes. When the calculation is whole, the Webina net app will transfer to the "output" tab (defined underneath) in which users can visualize the docking consequences.
Existing Vina output tab
The "existing Vina output" tab allows users to load and visualize the outcomes of previous Webina and Vina runs, without having to rerun the calculations. Users should specify the existing receptor and Webina/Vina output file they desire to visualize. They also can optionally specify a recognised-pose ligand document for reference. Users who wish to test the internet app without imparting their personal files can click on the "use example files" button. Otherwise the "load files" button will open and visualize the desired documents.
Output tab
The "output" tab allows customers to visualise their Webina docking effects. The same tab also displays the output of any previous Webina/Vina calculations that the user specifies thru the "existing Vina output" tab.
Visualization
The "visualization" subsection makes use of 3Dmol.Js to show the receptor and docked molecule in cartoon/surface and sticks illustration, respectively. If the user has specific an acknowledged pose ligand report, that pose is likewise displayed in yellow sticks. Like Vina, Webina predicts numerous poses in step with input ligand. A table beneath the visualization viewport lists each pose collectively with related data which include the docking score. Clicking on a desk row updates the three-D view with the required pose so customers can without difficulty observe all predicted poses (Figure 5).

Figure 5: Visualization after docking.
Output files
The "output files" subsection shows the text contents of the Webina output files. A related "download" button lets in customers to easily shop the ones files.
Run Vina from command line
Similar to the "input parameters" tab, the "output" tab also consists of a "run Vina from command line" subsection. This subsection makes it smooth for users to reproduce Webina's results the use of stand-on my own Vina. It also reminds customers what parameters they decided on to generate the displayed Webina output
Start over tab
The "start over" tab presentations an easy button that lets in the consumer to restart the Webina app. A caution message reminds the user that they will lose the results of the cutting-edge Webina run except they've stored their output documents.
The 18 tripeptide derivatives have been installation with the aid of utilizing at the net tool Webina in opposition to 4FGW protein. The docking protocol became demonstrated by means of considering RMSD (Root Mean Square Deviation) <1Ao between goal protein and preferred ligand. The peptide derivatives were evaluated for anti-diabetic pastime, the A2, A7, A9, B1, B2 and B3 have been displaying giant interest, A3, A4 were showing good activity and A1, A6 and A8 have been showing mild hobby and the results of docked peptide derivatives were proven within the Table 1.
| Compound code | Structure | Docking score (Kcal/mol) |
|---|---|---|
| Metformin | 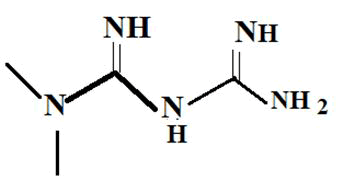 |
-4.4 |
| A1 | 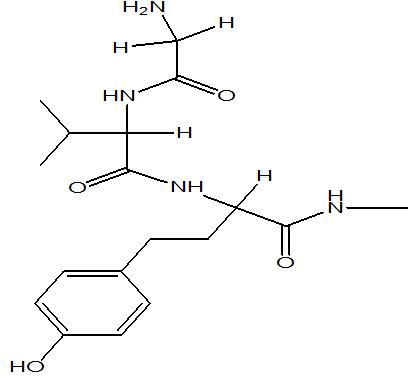 |
-7.4 |
| A2 | 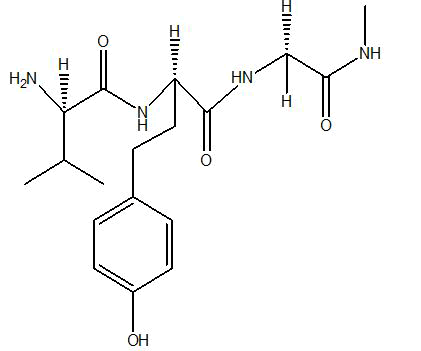 |
-5.6 |
| A3 | 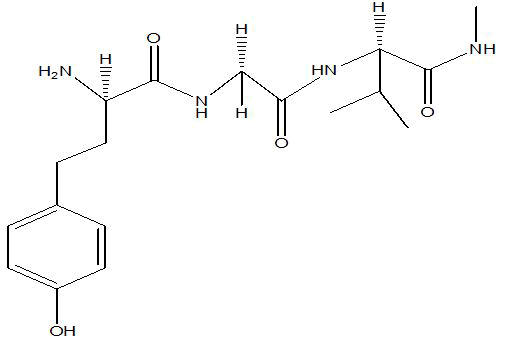 |
-4.6 |
| A4 | 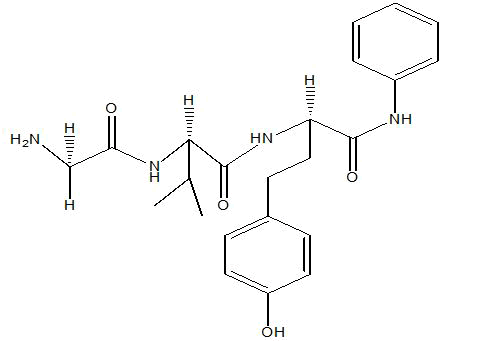 |
-3.4 |
| A5 | 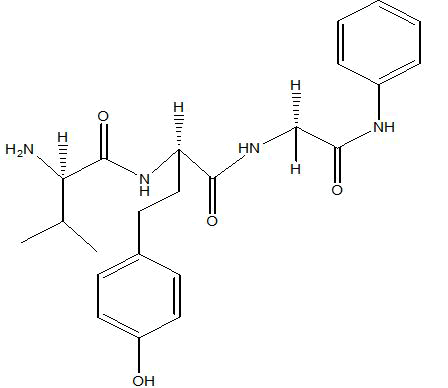 |
-3.4 |
| A6 | 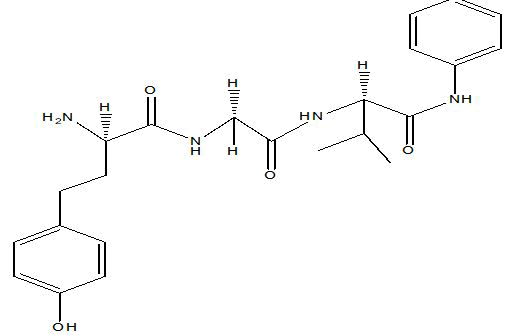 |
-2 |
| A7 | 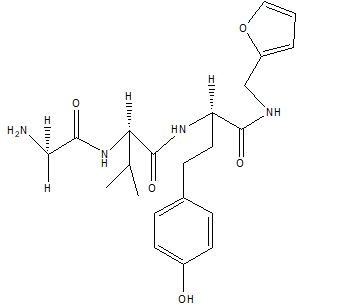 |
-8.6 |
| A8 | 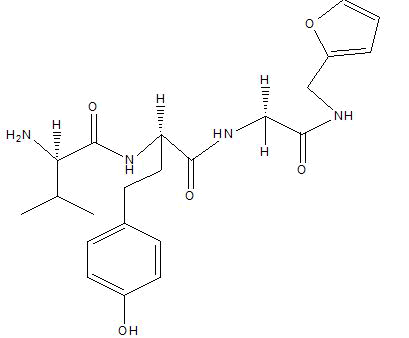 |
-7.6 |
| A9 | 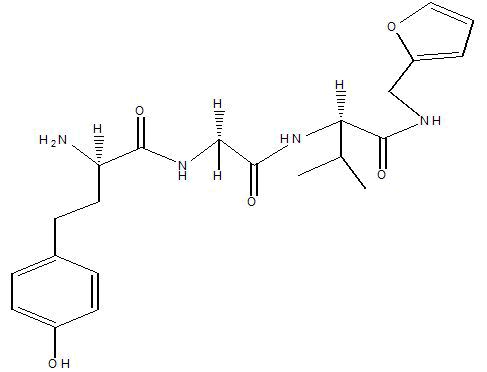 |
-1.6 |
| B1 | 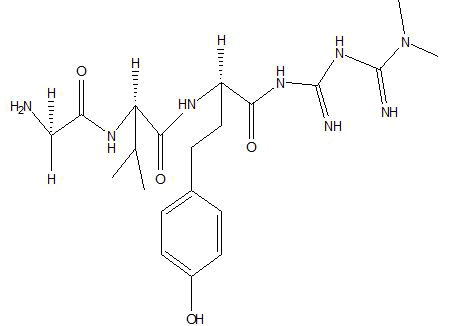 |
-6.6 |
| B2 | 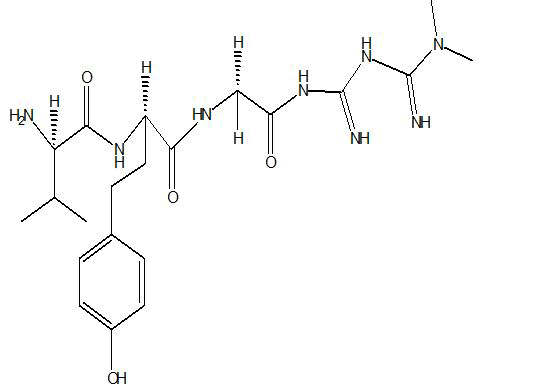 |
-7.4 |
| B3 | 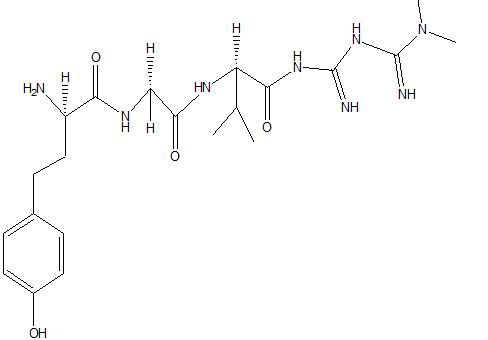 |
-5.7 |
| B4 | 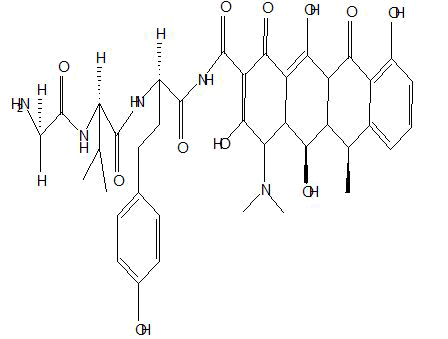 |
-6.1 |
| B5 | 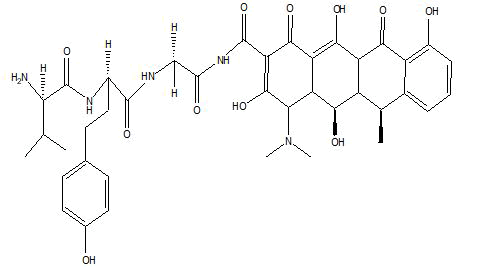 |
-4.8 |
| B6 | 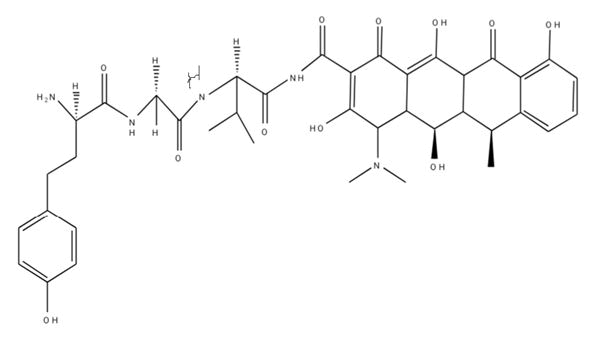 |
-5.6 |
| B7 | 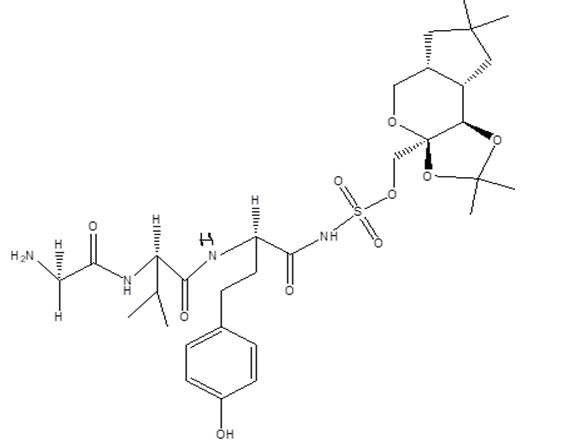 |
-6.4 |
| B8 | 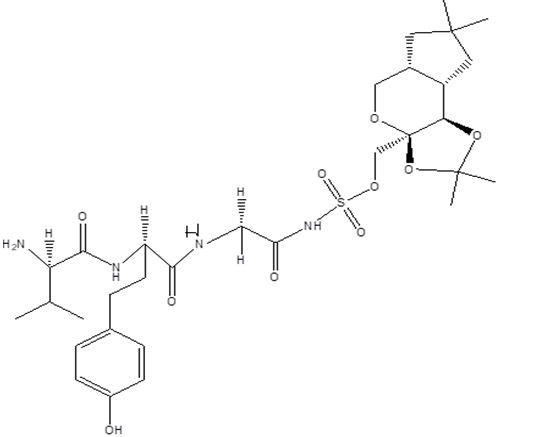 |
-7.2 |
| B9 | 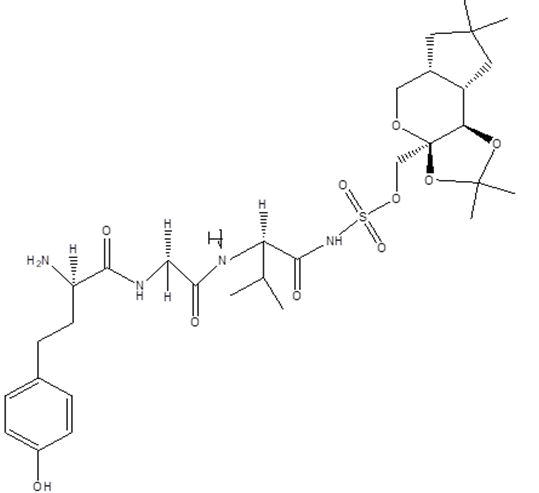 |
-6.5 |
Table 1: Docking score of synthesized peptide derivatives.
A total 18 synthesized peptide derivatives had been completed for molecular docking research with 4FGW protein thru using Webina internet server. The docking protocol became confirmed with the aid of considering RMSD (Root Mean Square Deviation) <1Ao amongst purpose protein and popular ligand (which docking score is -4.4 Kcal/mol). The peptide derivatives had been evaluated for anti-diabetic hobby the A2, A3, B1, B2 and B3 with docking rankings -5.6, -4.6, -4.8, -5.8 and -5.4 Kcal/mol respectively were showing precise interest and the effects of docked tri-peptide derivatives had been proven in the table number one. Total 18 designed tri-peptide derivatives docking rankings of compounds compared with preferred. The A2, A3, B1, B2 and B3 were displaying precise pastime. From the outcomes it changed into concluded that A1-A9 are displaying moderate activity, B1-B3 are displaying proper antidiabetic pastime.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Govindarajan R, Lakshmi VN (2023) Molecular Docking of Synthesized Tri-Peptide Derivatives by Using WEBINA 1.0.3. Drug Des. 12:252.
Received: 27-Dec-2022, Manuscript No. DDO-22-21180; Editor assigned: 29-Dec-2022, Pre QC No. DDO-22-21180 (PQ); Reviewed: 12-Jan-2023, QC No. DDO-22-21180; Revised: 16-Mar-2023, Manuscript No. DDO-22-21180 (R); Published: 29-Sep-2023 , DOI: 10.35248/2169-0138.23.12.236
Copyright: © 2023 Govindarajan R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.