Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Short Communication - (2022)Volume 11, Issue 4
In this study I have approached through in-silico method or reverse vaccinology taking advantage of the genome sequence of hepatitis G virus. It serves its benefit of identifying antigens seen by both conventional as well as discovering any novel antigen. This peptide candidate can serve a triple purpose of hepatitis C vaccine, hepatitis G vaccine and HIV management addition. 89.2% of the residues were in the favoured region of Ramachandran plot. These points make it favourable for in-vitro trials and further refinement. Because of the high similarity of hepatitis C genome to hepatitis G genome, it is highly probable that this peptide sequence might act as both hepatitis C and hepatitis G vaccine. Patients with past or current HGV infection have higher CD4+ lymphocyte counts and better AIDS-free survival rates. This peptide sequence might cause a breakthrough in the treatment of HIV without exposing them to develop hepatitis.
Hepatitis G; Vaccine; Hepatitis C; HIV; Peptide; Reverse vaccinology
With technological advancement in the field of immunology these studies have become easier and more accurate [1,2]. This peptide candidate can serve a triple purpose of hepatitis C vaccine, hepatitis G vaccine and HIV management addition. The procedure used in this study is entirely based on two previous studies [3,4]. It will not be repeated here but is summarized below in Figure 1 after obtaining necessary permission from its authors.

Figure 1: Methodology summary from a similar previous study.
Reproduced with permission from the authors.
The modifications done in this study is as follows
The genome sequence of hepatitis G virus was taken from NCBI virus database [5] (accession Number: NC001710) Polyprotein precursor protein of this genome is used in this study. Docking of final Peptide sequence was done with tertiary structure of toll-like receptor 7 obtained from Protein Data Bank [6] (PDB ID:5GMF).
The final multi-epitope sequence formed after performing step 1 and 2 of methodology comprised of 214 amino acids
GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCC RRKEAAAKAVEAGVT WYAAYLLDFVFVLLAAYVTDAVAAIQAAYDVALETELYGPGP GWPLYQAGLAVRP GKSGPGPGAASYLMGLGVGGNAQGPGPGPLYQAGLAVRP GKSAGPGPGAVFFSGL APLRMHPDGPGPGASYLMGLGVGGNAQTGPGPGVFFSGL APLRMHPDV
The above sequence distribution is as follows
GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCC RRK EAAAK (The above sequence is beta-defensin adjuvant sequence with EAAAK linker) AVEAGVTWY AAY LLDFVFVLL AAY VTDAVAAIQ AAY DVALETELY GPGPG
(Polyprotein precursor CTL epitopes linked to each other by AAY linker with highest immunogenicity and at the end with GPGPG linker to HTL epitopes).
 >
>
The antigenicity prediction as in step 3 by Vaxijen server predicted it to be a probable antigen with score 0.462. The allergenicity prediction as in step 3 by Algpred server predicted it to be a non-allergen with score of -0.86908598 (positive predictive value is 0% and negative predictive value is 0%).
Physio-chemical properties as estimated by step 4 using ProtParam server gave the following results
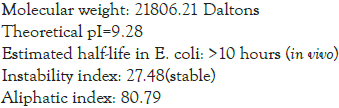
Grand average of hydropathicity (GRAVY): 0.096. The secondary structure of the final multi-epitope sequence was computed using PHYRE 2 server: 40% comprised of alpha-helix, 3% of beta-strand, 13% of trans membrane helix and 22% was disordered (Figure 2). The tertiary structure obtained from PHYRE2 server was subjected to refinement by Galaxy Rene tool which generated 5 models as follows in Table 1: Model 4 was chosen as the best tertiary structure of the sequence for further analysis. It was visualized using UCSF Chimera software (Figure 3) [7].

Figure 2: Secondary structure of vaccine sequence. Note: Disordered (22%),
Disordered (22%),  Alpha helix (40%),
Alpha helix (40%),  Beta strand (3%),
Beta strand (3%),  TM helix (13%)
TM helix (13%)

Figure 3: Model 4 tertiary structure.
Ramachandran plot analysis by RAMPAGE server Figure 4 gave the following result: Number of residues in favoured region (~98.0% expected): 189 (89.2%); Number of residues in allowed region (~2.0% expected): 15 (7.1%); Number of residues in outlier region: 8 (3.8%). The predicted B-cell linear epitopes were calculated using Ellipro suite (Table 2 and Figure 5). Toll-like receptor 7 was docked with the final model by PatchDock server and top 10 results were refined using FireDock server. Solution number 2 was the most favourable binding conformation with global energy at -4.41 and 0.00 repulsive Vander Waal forces. The docked model was visualized using UCSF Chimera (Figure 6).

Figure 4: Ramachandran plot analysis of the final vaccine tertiary structure. Note:  General/Pro/Proline Favouerd,
General/Pro/Proline Favouerd, General/Pre-Pro/Proline Allowed,
General/Pre-Pro/Proline Allowed,  Glycine Favoured,
Glycine Favoured,  Glycine Allowed.
Glycine Allowed.

Figure 5: Epitope score chart from Ellipro suite.

Figure 6: Peptide model (red) docked with Toll-like receptor 7 (blue).
The protein sequence is predicted to be antigenic as well as non- allergic, hence proving its advantage of not producing any harmful hypersensitivity reaction in the body. It is basic in nature and has low molecular weight hence suitable for any route of administration except oral. It’s half-life in E. coli is >10 hours, hence can easily be cultured and extracted. It is thermally stable as indicated by instability index. It has various B-cell epitope stimulating site and molecular docking with toll like receptor TLR-7 shows that it binds easily it without any repulsive Van der Waal forces. Toll- like receptor 7 which induces immune response against ss-RNA organisms will elicit an immune response against this sequence considering it be an active virus and thus fulfilling its purpose as a vaccine. 89.2% of the residues were in the favoured region of Ramachandran plot. These points make it favourable for in-vitro trials and further refinement.
All these studies were on web-tool prediction servers designed for such type of studies. Because of the high similarity of hepatitis C genome to hepatitis G genome, it is highly probable that this peptide sequence might act as both hepatitis C and hepatitis G vaccine [8]. Patients with past or current HGV Page 5/12 infection have higher CD4+ lymphocyte counts and better AIDS-free survival rates [9-11]. This peptide sequence might cause a breakthrough in the treatment of HIV without exposing them to develop hepatitis. However, since they work on growing databases, these cannot give a complete surety for success in future stages. Along with the advantage of the study, there are some limitations. 22% of the predicted secondary structure is disordered. Instead of 98% proteins being in the favourable region of Ramachandran plot only 89.2% of them are present. Advanced molecular dynamic simulations were not performed like RMSD (root mean square deviation).
These disadvantages need to be overcome with better resources but these early results do serve as a guiding path to build future work upon it. This study has highlighted a potential candidate fulfilling its purpose as hepatitis C vaccine, hepatitis G vaccine and HIV management addition. In-depth studies and refinement might serve to be successful since it has very good results at such an early stage. It is needed to be validated experimentally.
Conflict of interest
The author declares no conflict of interest.
Nil.
Not required.
[Crossref]
[Crossref]
Citation: Sharp K (2022) Multi-Epitope Peptide Sequence in-silico Construction from HGV Genome . Enz Eng. 11:188.
Received: 01-Jun-2022, Manuscript No. EEG-22-17695; Editor assigned: 03-Jun-2022, Pre QC No. EEG-22-17695 (PQ); Reviewed: 21-Jun-2022, QC No. EEG-22-17695; Revised: 27-Jun-2022, Manuscript No. EEG-22-17695 (R); Published: 04-Jul-2022 , DOI: 10.35841/2329-6674.22.11.188
Copyright: © 2022 Sharp K. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.