Fungal Genomics & Biology
Open Access
ISSN: 2165-8056
ISSN: 2165-8056
Review Article - (2015) Volume 5, Issue 1
Keywords: Fungi, Metagenomics, Host microbe interaction, Pathogenicity, Dysbiosis
The advent of metagenomics is clearly demonstrating how we are walking holobiomes [1], made of the genes of our genome and those of our metagenome [2–6]. This concept implies that the genes governing the interaction of the microbiota and the host are key determinants of health and disease. Darwinian selection is therefore acting on a complex community of organisms interacting at the organismal level to build organs, bodies, communities. Deep sequencing allowed to characterize in detail these populations, revealing that the human microbiota differs radically at different body sites and among individuals [2,3]. Historically microorganisms that colonize us at birth come from our mothers’s vaginal tract and breast milk [7–9] . During the development of our immune system we are then exposed to microbial communities that enter our body through water, food, and the environment where we grow adult. The origin of differences between individuals is still debated, however, potentially reflecting distinct colonization early in life [10], type and availability of nutrients and different dietary regimens [11,12], and different environmental exposures to microorganism [13], such as antibiotic use [14]. The history of our encounters with microorganims shapes our immunological make-up, but on the evolutionary time scale, our immune system has evolved to tolerate certain microbes and attack, get rid of others. Nowadays, bacterial genomic sequence data have generated novel hypotheses about species and community structures underlying human disorders of immune origin [15]. The application of microbial ecology concepts is ultimately leading to the conclusion that health and disease can be understood only understanding how the symbiotic interactions between microbes and human organs harmonically integrate [1].
Due to the greater abundance of bacteria over fungi, investigators have mostly focused on the prokaryotic component of the microbiota, but fungi are increasingly being recognized to have a role in defining these communities and interact with our immune system [16].
Fungal eukaryotes have been implicated as causative agents of diseases such as Inflammatory Bowel Syndrome (IBS) (Blasto-cystis), Inflammatory Bowel Disease (IBD) (fungi), and “leaky gut” syndrome (Candida) [16–18]. These diseases, which have become prevalent in western populations, are due to complex changes in the microbial community. Elucidating community-wide changes, rather than presence or absence of specific taxa, will be crucial to understanding the cause and potential treatment in these many-faceted poly-microbial diseases [19].
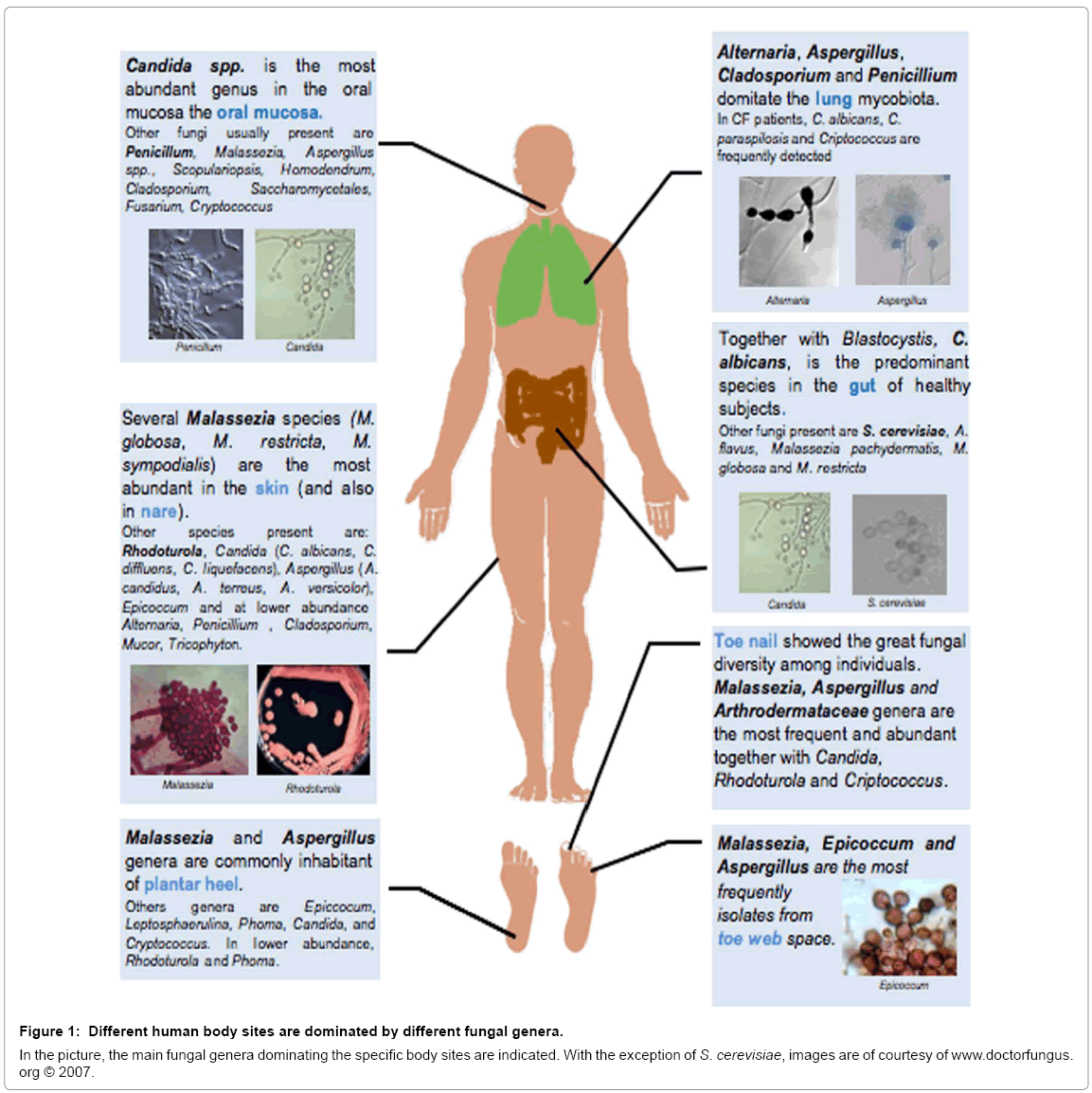
More and more information on the incidence and diversity of fungi in humans are accumulating, and the primary aim of this review is to convince you that the current studies are just describing the tip of the iceberg. We will geographically describe the body map of our fungal communities (Figure 1) stressing the importance to study mycobiota in the context of the bacterial counterpart.
Investigating the mycobiota: target and untargeted metagenomics
Comprehensive study of the eukaryotic component of the human microbiota is just beginning and lags far behind our understanding of the bacterial communities. Culture-based methods relied on enumerating the cultivable part of mycobiota, not capturing overall community structure and its spatial and dynamic properties. And reports using NGS suggest diverse fungal communities in humans (as described below). High-throughput sequencing technologies made metagenomics (i.e the direct sequencing of fungal DNA) the main method for characterizing the mycobiota.
There are two main approaches used in metagenomics, untargeted metagenomics and targeted metagenomics [20]. The untargeted approach sequences all the DNA content of a sample without amplification using mainly the Illumina/Solexa sequencing platforms [21]. The principle is the sequencing-by-synthesis technology, but it is based on the use of reversible terminator nucleotides labelled with a different fluorescent dye depending on the type of nucleotide [22]. The most recent advancements in ecological metagenomics are indeed going to use untargeted approaches sequencing the entire DNA content of a sample, the actual utility of these futuristic approaches, already of common use for human metagenomics, largely depends on the number of sequenced and annotated genomes of environmental microbes, an aspect that currently provides the greatest challenge for the wider scientific community working on environmental microbiology [23]. Application of untargeted methods to investigate fungal communities is extremely difficult since the relative abundance of bacterial cells reduces the overall number of eukaryotic or fungal reads.
The targeted approaches uses the sequence of molecular clock genes to infer the phylogenetic structure of the microbial community. Predominant among the targeted approaches, is ribosomal RNA, rRNA phylotyping [24]. The success of this technique is based on the enormous database of rRNA gene sequences (more than 200,000) that have been collected for reconstructing the universal Tree of Life [25]. Targeted approaches are currently the method of choice for the investigation of fungal populations. The most sensitive and affordable approach for fungal targeted metagenomics is to sequence a small variable region of fungal genomes like internal transcribed spacer regions, 18S small-subunit ribosomal DNA (rDNA), 28S large-subunit rDNA. The ribosomes of eukaryotic organisms are composed of two structural subunits, 60S and 40S. The 60S subunit contain three different types of rRNA fragments: 28S, 5.8S and 5S rRNA. The 40S has a single 18S RNA fragment. The genes coding for the different fragments of rRNA are repeated genes, constituting the nucleolus organize region (NOR). Within the chromosome are clustered into structural units separated by non-coding spacer sequences called NTS (non-transcribed spacer) and IGS (intergenic spacer). Each unit consists of a structural and external transcribed spacer (ETS), located at the 3 ‘end of the 18S gene, two internal transcribed spacers (ITS), situated respectively: the ITS1 between the gene 18S and 5.8 S gene, the ITS2 between the 5.8S 3 ‘end and the 28S gene and 5S gene is separated from the 28S gene from another non-coding spacer sequence. ITS1 region, such V3-V5 region of the 16S, are hypervariable regions, so their sequencing allows us to identify fungi at the species or genus level, and in the case of a metagenomic analysis, to understand the composition of the fungal communities present in a particular habitat. In a number of studies the co-sequencing of fungal 18S rDNA/ITS and bacterial 16S rDNA sequences resulted the optimal method for the investigation of the biodiversity and function of the global microbiota which exists in each part of the human body [26,27]. Recently co-sequencing of 18S rDNA and ITS has been proposed as more suitable for fungal classification. Indeed the results’ quality depends on having a comprehensive, well-annotated database of fungal sequences with which to compare the sequencing data. Whereas several excellent databases have been developed for identifying bacterial 16S rDNA sequences, fungal sequences databases are still in their infancy, where less than 1% of fungal species are currently represented [28]. Analyzing NGS data requires even more new computational tools, such as co-occurrence networks, protein-protein interactome [29] and pipelines for comparing large numbers of samples, enabling, in the next future, the patterns in these data to be elucidated. Furthermore, fungal taxonomy is currently improving as fast as the different fungi are characterized. Sequencing and annotating much more fungal genomes is required to properly delve into the biology and the interactions that make the complexity of their population and community structure. How changes in the fungal and bacterial community structure leads to health or disease can be understood only in the context of the evolutionary forces shaping their adaptation to the host.
Despite the existing limitations the initial application of metagenomics to fungal communities has produced in the past three years more information of that obtained in the previous thirty. The information summarized in Figure 1 is indeed going to rapidly evolve with the exponential increase of community level genome-wide surveys of the microorganisms inhabiting the various microenvironments of the human body (i.e. gut, skin, oral mucosa and urogenital tract), their environmental reservoir, and the human populations living in different geographic regions.
The host and its double: being a commensal or a pathogen is a matter of dysbiosis
Pathogens are typically considered as discrete causative agents in virulence studies, even if they exist within a rich milieu of other microbial species, as well as host factor, that can influence pathogenicity. Thus, disease could derive from dysbiosis of the microbial community that refers to a condition with microbial imbalances on or inside the body. This paradigm requires that diseases of microbial origin must be investigated within the context of their microbial community, host factors and immunity. Genome-wide surveys of the microorganisms inhabiting the different microenvironments of the human body (i.e gut, skin, oral mucosa and urogenital tract) [30], soil [31], and population living in different geographic regions [12,13] have suggested the presence of strong selective control on the microbial communities that can colonize and persist in particular environments. Thus, understanding the prevalence and distribution of microbial eukaryotes, in addition to prokaryotic microorganisms, in the human body, may have large consequences for human health.
While the focus is generally, in terms of study effort, genome sequencing, etc., on pathogens or conditional pathogens (i.e. their pathogenic potential is unleashed upon failure from the immune system to keep them at bay), most microbial resident eukaryotes do not cause infections, being either beneficial or commensal. Commensal microbes significantly shape mammalian immunity, both at the host mucosal surface and systemically [32], controlling unexpected microbial burden and growth.
However, fungi belonging to commensal genera such as Candida and Malassezia show a clear potential to infect the host when they become abundant or infect immunocompromised subjects.
At mucosal surfaces, those opportunistic fungi are controlled by the normal microbial flora, the epithelium and its innate immune system: this tight control allows to refer to them as part of the normal mycobiota. Although the commensal stage is frequently reported as inoffensive to the host, it is likely that this stage is highly regulated by a continuous or transient cross-talk between the fungus and the host immune system [33]. The latter maintains homeostasis with resident microbial populations, ensuring the balance between tolerogenic and pro-inflammatory response. In parallel, commensal microbes significantly shape mammalian immunity both at the host mucosal surface and systemically [33,34] controlling un-expected microbial burden and growth.
Mucosal fungi modulate both the innate and adaptive immune responses. C. albicans is able to train the innate immunity towards other microorganisms, such as intestinal and skin bacteria [34–36]. Even S. cerevisiae, which presence was recently reported in the human skin [16,27,37,38] and in the gut microbiota (De Filippo et al., submitted) educates the host immune response to better cope a secondary infection (Rizzetto et al., submitted).
Indeed, the configuration of the commensal microbiota is being newly appreciated as complex and important for health, with increasing evidences suggesting a novel perspective on the role of non-pathogenic fungi in contributing to a correct immune homeostasis [39–41].
An increased understanding of the importance of microbiota in shaping the host’s immune and metabolic activities has rendered fungal interactions with their hosts more complex than previously appreciated. It is now clear that a three-way interaction between host, fungi, and microbiota drives the host-fungus relationship. The aryl hydrocarbon receptor (AhR) has a pivotal role in driving this connection linking tryptophan catabolism by microbial communities and the host’s own pathway of tryptophan metabolite production. Its activation by the commensal microbial community, it plays an antimicrobial role and an anti-inflammatory role - mediating the differentiation of regulatory T cells [42]. In addition to the evidence that via AhR activation Lactobacillus reuteri, contributes to mucosal resistance against the opportunistic pathogen Candida. albicans [43], an increased concentration of AhR ligands in the skin has been linked to the development of Malassezia-associated skin diseases [44,45].
Indeed, microbial dysbiosis predisposes either causally or consequently to a variety of diseases including cystic fibrosis CF [46], inflammatory bowel disease (IBD) [47], atopic dermatitis [48], and chronic mucocutaneous candidiasis (CMC) [49]. However, it has been unclear whether this variation is primary or secondary to an imbalanced bacterial microbiota.
Physical and quorum sensing interactions occur between fungi and bacteria. Those interactions can be antagonistic, synergistic, commensal or symbiotic and influence physical and physiological characteristics such as the mutual morphology, behavior and survival including response to anti-microbial agents [50]. It has been shown that in the mouse gastro-intestinal gut, commensal fungi co-exists in the same patches with bacteria [51]. Quorum-sensing molecules are often the basis of the interaction between fungi and bacteria. For example, C. albicans can inhibit the virulence of P. aeruginosa through farnesol, while P. aeruginosa can affect the growth, morphology and virulence of C. albicans through the virulence factors pyocyanin and phenazines [52–54].
Moreover, germ-free mice are highly susceptible to Candida infection [55], and antibiotic treatment supports colonization by and robust expansion of fungal species in the mouse gut [56–58], reaching the 99% of the total intestinal microorganisms [58]. This was mostly due to an expansion of Candida species, which was the only genus that was found, by deep sequencing, in the feces. The same occurs in humans: prolonged antibiotic treatment predispose to Candida infections [59,60].
Moreover, a recent study has demonstrated that after a broad spectrum antibiotic treatment with cefoperazone, the colonization with C. albicans strongly altered the restoration of the bacterial communities after treatment [61]. The presence of the fungus shifts the bacterial proportion: Bacteriodetes and Lactobacillaceae decrease in size, while Firmicutes populations remained unchanged.
Collectively, the eukaryotic and prokaryotic communities are kept in equilibrium by mutual interactions that include production of immune modulating molecules, helping to accommodate fungi, either commensals or ubiquitous, within the immune homeostasis and its dysregulation. Building a large database of co-occurring eukaryotic and bacterial communities will allow one to test the hypothesis that changes in the bacterial community are responsible for the variance in the pathogenicity of eukaryotic microorganisms or vice versa.
Fungi inhabiting the skin surfaces
The skin represents the primary interface between the host and the environment. Deep sequencing approaches have demonstrated that the healthy skin contains a unique bacterial and fungal microbiota [30,38,62–64]. The composition of this microbial community depends on physiologic attributes and topography of the skin, with certain patterns being associated with moist, dry or sebaceous microenvironments [30,62]. Culture-dependent approaches have identified many commensal fungi that are present on the skin, with Malassezia as the most common genus, followed by Penicillium and Aspergillus [38,62,65]. Other fungi, such as Alternaria, Candida, Rhodotorula and Cladosporium, have also been cultured but with a lower frequency [38,64,65]. Culture-independent sequencing studies have confirmed that the genus Malassezia is most commonly represented on healthy human skin across different body sites [38,48,66]. Eleven body sites were found to be dominated by Malassezia fungi but by contrast [38], foot sites (plantar heel, toe nail, and toe web) exhibited the greatest fungal diversity (40-80 genera), with the presence of a wide range of fungal genera (i.e Rhodoturola, Debaromyces, Cryptococcus, and Candida), of which Candida species were mostly represented by C. tropicalis, C. parapsilosis and C. orthopsilosis. Changes in the fungal microbiota of the scalp that accompany dandruff have been also examined [67]. While fungi of the Ascomycota dominated in both healthy and dandruff patients, fungi of the Basidiomycota phyla (which includes Malassezia) were significantly increased in dandruff-afflicted scalps. Specific genera that were increased in patients with severe dandruff included Filobasidium, which jumped to 94% of the total basiomycetes, and Malassezia (5%), which represents a twofold increase over healthy samples. By contrast, healthy scalps were dominated by basiomycete fungi of the genus Cryptococcus [67].
Cutaneous inflammatory disorders such as psoriasis, atopic dermatitis (AD), and rosacea have been associated with dysbiosis in the cutaneous microbiota [68–71].
To characterize that dysbiosis, Zhang and coworkers used an rRNA gene clone library of 3647 clones to identify 58 fungal species and 7 unknown phylotypes from AD patients and healthy individuals [48]. As expected, Malassezia species were predominant, accounting for 63–86% of the clones identified from each subject. Overall, AD patients showed an increase diversity of the non-Malassezia yeast communities with respect to healthy subjects. C. albicans, C. diffluens, and C. liquefaciens as well as the filamentous fungi Cladosporiumn gi spp. and Toxico cladosporium irritans were detected in AD samples but were seldom detected in healthy samples [48].
An exemplary recent publication [36] has added another fundamental contribution to the role of skin microbiota in activating and educating host immunity, shedding new light on the interplay between the immune system and microbiota. The authors studied patients with hyper IgE syndrome, a primary immunodeficiency disease (PID) due to STAT3 deficiency, and compared the bacterial and fungal skin microbiota at four clinically relevant sites representing the major skin microenvironments [36]. The patients exhibited increased ecological permissiveness, characterized by an altered bacterial community (showing a colonization with Clostridium spp and Serratia marcescens) and an increased fungal diversity with augmented abundance of Candida and Aspergillus species. This increased representation of opportunistic fungi was concomitant with a decrease in the relative abundance of the common skin fungus Malassezia [36].
Analyses of the co-occurrence of specific bacteria and fungi have suggested that the two communities might interact in the skin mucosal surface. The intra-kingdom interaction seems to occur with Malassezia globosa and most genera of Firmicutes and Actinobacteria in both controls and STAT3-HIES patients with a correlated reduction of Malassezia restricta. In patients’ group, streptococci, coryneforms, and S. aureus were uniquely correlated with the presence of Candida, Aspergillus and Cryptococcus.
The skin microbiota investigation provides an important step toward understanding the interactions between pathogenic and commensal fungal and bacterial communities, and how these interactions can result in beneficial or detrimental (i.e., disease) outcomes.
Fungi inhabiting the oral mucosa
The oral microbiota is a critical component of health and disease. Disruption of this community has been proposed to trigger or influence the course of oral diseases, especially among immunocompromised patients [72]. Metagenomic sampling of individual sites within the oral cavity shows that there are probably hundreds of different microbial niches [73–76]. The fungal component of the oral microbiota, however, has been only recently characterized. The most comprehensive study to date on the fungal microbiota of healthy mouth found that the distribution of fungal species in the mouth varied greatly between different individuals. This multi-tag pyrosequencing approach showed that the oral cavity hosts to >75 different fungal genera with Candida, Cladosporium, Aureobasidium, Saccharomycetales, Aspergillus and Fusarium being among the most common [27]. The remaining 60 nonpathogenic fungi detected in the oral wash samples represent species that likely originate from the environment in the form of spores inhaled from the air, or from material ingested with food. Thus, the presence of these microbes in the oral cavities of healthy individuals was not necessarily surprising, but the observation that transient colonization by environmental fungi may occur in the oral cavity (and upper airways) has potential implications for hypersensitivity diseases.
Using a different approach based on high-throughput sequencing analysis of ITS1 variable region, a subsequent study reported that Malassezia spp. and Epicoccum are also abundant [77] in the saliva of healthy subjects.
The presence of pathogenic fungal isolates in the oral cavity of healthy individuals is quite unexpected and the clinical relevance is unknown. It is possible that the presence of a given fungal isolate in an individual could be the first step toward predisposing that individual to opportunistic infections. The pathogenicity of the fungi in the oral environment may be controlled in healthy individuals by other fungi or other member of the oral community, as well as by the functional immune system, suggesting that interdependent cross-talk may exist between constituents of the oral mycobiota. Surveying the oral microbiota of 12 HIV-infected patients and 12 healthy subjects showed that the core oral bacterial microbiota comprised 14 genera, of which 13 were common between patients and healthy subjects [78]. In contrast, the core oral mycobiota differed between HIV-infected and uninfected individuals, with Candida being the predominant species in immunocompromised patients. Increase in Candida colonization, particularly C. albicans, was associated with a concomitant decrease in the abundance of Pichia - a resident oral fungus, suggesting an antagonistic relationship. Indeed, Pichia inhibits growth of pathogenic fungi; such as Aspergillus and Candida by inhibit the ability to adhere, germinate, and form biofilms [78].
Oral Candida colonization is a known risk factor for invasive Candidiosis [79]. Similarly, periodontal disease is associated with rheumatoid arthritis [80] and atherosclerosis [81], suggesting that bacterial and fungal microbiota from the oral cavity may contribute to the development of certain human diseases. According to that, Candida albicans and Streptococcus spp. can form a mixed biofilm with antimicrobial antagonism by a complement of adhesin-receptor interactions involving two families of streptococcal multifunctional polypeptide adhesins and bacterial cell wall polysaccharide [82,83]. This physical interaction results in a synergistic increase of their invasiveness in the oral and upper gastro-intestinal tract mucosa in vivo [82].
Fungi inhabiting the lung mucosa
Although the lungs are constantly exposed to oral and environmental fungi [84], the lower respiratory tract (trachea, bronchi and pulmonary tissue) was thought to be sterile when healthy.
However, if the respiratory tract epithelium becomes damaged, as in bronchitis or viral pneumonia, the individual may become susceptible to infection by pathogens, such as Aspergillus, descending from the nasopharynx (upper respiratory tract). The normal bacterial part of microbiota has been addressed more deeply than its fungal counterpart. It consists of 9 core genera: Prevotella, Sphingomonas, Pseudomonas, Acinetobacter, Fusobacterium, Megasphaera, Veillonella, Staphylococcus, and Streptococcus [85,86] but few data exist about the fungal microbiota of the lungs, with the exception of Pneumocystis spp. In a recent study by Charlson and collegues, the fungal microbiota of the mouth and lungs in select healthy and lung transplant recipients was analyzed by ITS-based pyrosequencing [87]. The patients’ fungal microbiota of the oral cavity was dominated by Candida, while bronchoalveolar lavage showed detectable Candida spp., Aspergillus spp., or Cryptococcus spp. This mycobiota composition likely depends on the antibiotic and immunosuppressant use supporting the notion that heathy host defense, and perhaps some complex interplay between lung-colonizing microbial communities and invading pathogens, play a major role in keeping fungal colonization extremely low in the lungs.
Cystic fibrosis (CF) provides an important example of the need to enhance our knowledge of the composition of the microbial community in order to improve management of patients susceptible to pulmonary infections. Using ITS2 and 16 rRNA pyrosequencing, Delhaes and coworkers [46] extensively explored the diversity and dynamics of fungal and prokaryotic populations in the lower airways of CF patients. The authors identified 30 genera, including 24 micromycetes, such as Pneumocystis jirovecii or Malassezia spp, and 6 basidiomycetous fungi. Among the organisms identified, filamentous fungi belonging to the genera Aspergillus and Penicillium had previously been suggested as pathogens in CF patients [88]. C. albicans and C. parapsilosis were also recently described as colonizer organisms of CF patients [89,90].
CF microbiota was characterize by a large proportion of anerobes and in particular by Pseudomonas aeruginosa, which was more likely to be observed in association with Candida. albicans than with Aspergillus. fumigatus [46]. This can be explained by the fact that colonization of C. albicans in rat lungs prior to Pseudomonas spp. exposure may increase the incidence of Pseudomonas-related pneumonia, because C. albicans can impair the local host immune response by inhibiting reactive oxygen species production by alveolar macrophages [91].
Keeping in mind the co-occurrence of the microbial communities transiently or persistently colonizing the lung [92,93], a modification in lung mycobiota may result from a primary dysbiosis of bacteria. Indeed, the causative or the correlative relation between changes in lung mycobiota and disease onset needs to be proven by expanding the number of samples and moving forward the study from the species to the strain level.
Fungi inhabiting the vaginal mucosa
Vagina hosts a set of microbial occupants that, if not properly contained, can cause pathologies. While yeast infections are common, epidemiological and mycobiome studies reveal a more diverse population of resident fungi than has been previously appreciated [94–97] . These studies indicate the presence of 11–20 different genera, with commonly detected fungi including Candida (C. albicans, C. glabrata, C.parapsilosis, C. tropicalis, and C. krusei), Saccharomyces, Aspergillus, Alternaria and Cladosporium. Changes in fungal diversity were found to be associated with immuno deficiencies [97], diabetes [95], allergic rhinitis [96] and recurrent vaginal candidiasis[96]. Fungal growth is controlled both by host immune system and the competition with the local bacterial communities. Lactobacilli species are the dominant microorganisms in the healthy vaginal microbiota [98], which lactic acid production helps in maintaining an acid environment thus impeding fungal growth [99,100].
Fungi inhabiting the gut
The human gastrointestinal tract is known to contain a variable fungal microbiota, but the phylogenetic characteristics of those fungal microorganisms and their specific roles as part of the gastrointestinal tract (GI) ecosystem have not yet been studied extensively. Whereas initial cultured dependent surveys only suggested that human and mouse GI are populated by diverse fungal microbiota, more recently, high-throughput sequencing approaches have been used to in depth and quantitatively explore the fungal communities that populate both the human and mouse gut. These studies have shown that the gut host up to 50 genera of fungi with Candida, Saccharomyces and Cladosporium species being particularly common [51,101]. An example of the large variability of the human gut mycobiota was provided by a study of four children and their respective mothers, which reported that infants harbor Saccharomyces spp. as opposed to Candida as the most frequent fungal species (36%) [102]. A recent study conducted on Wayampi Amerindian community, showed a high diversity among yeast species, with a prevalence of S. cerevisiae over Candida species [37], suggesting a role for this fungus in gut immune homeostasis.
Whether S. cerevisiae is present at birth remains to be elucidated. It is possible that yeasts simply reach the gastrointestinal tract through food. Alternatively, it is possible that differences in fungal colonization are related to differences in the genetic makeup of the host or differences in gut permeability. A few studies have been conducted to examine fungal community dysbiosis in chronic disease, including IBD [16,51]. Studies of the mycobiota in a murine model of induced colitis highlighted the importance of gut fungal communities in contributing to the boost in intestinal inflammation seen upon DSS treatment [51], with a marked increase in the abundance of C. tropicalis observed during active colitis. These studies are the first steps toward clarifying the role of the gut mycobiota in intestinal inflammation, and may help explain the increased serum levels of anti-S. cerevisiae antibodies (ASCA) in CD patients [103].
A more recent study on 19 patients suffering from active Crohn’s disease CD investigating the mucosa-associated and fecal fungal microbiota showed that the fungal richness and diversity were higher in the inflamed mucosa compared with the non-inflamed mucosa. The predominant fungal composition of CD mucosa and feces are being characterized by expansion in the proportions of Candida spp., Gibberella moniliformis, Alternaria brassicicola, and Cryptococcus neoformans,Aspergillus. clavatus, and C. neoformans. The species richness and diversity of the mucosal fungal community were associated with the expression of TNFα, IFNγ, or IL-10 and positively correlated with serum C-reactive protein and CD activity index [47]. Whether changes in gut fungal microbiota are the cause or the consequence of mucosal inflammation and disease activity of CD remains yet to be understood. Additionally fungal role in health and disease could be that of favoring growth of certain bacterial species and limiting the growth of others. Lactobacillus spp. and C. albicans have antagonistic relationship inside the gut. Even if not able to completely eradicate C. albicans, the bacterium could inhibit the growth and virulence of C. albicans by the production of hydrogen peroxide and organic acids [104,105]. Moreover, commensal Lactobacilli produce the metabolite indole-3-aldeyde (IAld) by tryptophan metabolism, activating the Ahr mediated IL-22 response which in turns reduces colonization by C. albicans [43]. Indeed, Lactobacilli provide local protection and resistance to fungal infection.
Several reports suggest that diet is a main driver of gut fungal diversity. Studies in newborn mice and in human infants have suggested that bacterial communities in the gut are initially unstable but become more stable in early childhood and develop more modestly throughout adulthood [106,107]. However, this may not be the case with the gut mycobiota. One recent study demonstrated that fungal populations in the mouse gut displayed episodic variation over several months, although the bacterial community remained relatively stable [58]. This suggests that commensal fungal populations are more variable than those of bacteria and that they may be influenced by fungi in the environment. Diet is a constant and dynamic factor shaping mucosal immunity as well as the composition of resident microbial populations in the gut. Hoffmann et al. investigated the association of diet with fungal populations, analyzing 98 fecal samples [101]. Deep sequencing revealed 62 fungal genera and 184 species with generally mutually exclusive presence of either the phyla Ascomycota or Basiodiomycota. Moreover, the consumption of a plant based diet has been linked to an increase in gut colonization by Candida species, whereas the consumption of an animal-based diet facilitated the expansion of Penicillium species [11].
Thus, integrating information on the repertoire of the gut mycobiota in the context of the broader microbiota and developing functional tests to measure its role in shaping immune function is necessary to better understand the role of the microbial communities in sustaining human health. The development of a highly specialized symbiosis requires iterative sets of mutual adaptation between and among commensal microorganisms and their hosts. This requests integrating the microbial diversity surveys with the identification of specific metabolites that cab directly target the diverse immune function (i.e the IDO1- AhR axis) for the promotion of infection, its control and immune resistance.
Recent culture-independent surveys of eukaryotic communities reveal that, similar to bacteria, commensal fungi are an essential part of human ecosystems. Fundamental research on fungal communities of the human host will have to understand the role of diet and environmental changes in exposure to fungi. Globalization has meant diet standardization and elimination of microorganisms from food products, as well as an increased use of antibiotics. Both these factors are shown to alter significantly the bacterial composition of the gut microbiota of children living in the globalized world [12]. How globalization affects our exposure to dietary and environmental fungi is one of the most interesting paths of research yet to be explored.
The role of the mycobiota in the maintenance of health can only be understood only using a “systems level” integrated ecological approach, as opposed to an approach focused on specific, disease-causing taxa. Commensal fungal populations vary between body sites, vary over time and with disease, specifically interacting with the immune system. The molecular and cellular mechanisms that engage specific fungi during steady-state healthy conditions may be quite different from those that are engaged in diseased tissue or during active fungal invasion. Keeping in mind the ability of fungi to evade the immune system by subverting the host defense i.e by morphological and phenotypic transition it will be critical in the next future the need to functionally analyze the mycobiota at the strain level in its cross-talk with the immune cells and the bacterial counterpart, rather than simply counting its parts at the species level. This analysis will require developing strains specific markers as well as gaining an understanding of their population and community structures in the context of the evolutionary forces shaping their adaptation to the different body sites. Studying the role of fungi in contributing to immune homeostasis is indeed one of the major future challenges we face. Understanding this role could suggest how we could preserve the appropriate level of interaction between our immune system and fungi, and how this could help us developing fungal probiotics or maintaining the right balance between the right fungi and the right bacteria, to maintain a healthy immune function.
Future studies like in-depth sequencing of the meta-transcriptome of eukaryotes, during the interaction with the host immune system, will soon help in understanding host-microbes interaction and in exploiting this knowledge to prevent or treat fungal and inflammatory diseases.
We would like to thank Francesco Strati for the helpful discussion and Andrea Mancini for helping in figure’s editing.
Authors declare that any conflict of interests exists.
This work was supported from the Accordo di Programma Integrato “MetaFoodLabs” funded by the research office of the Provincia Autonoma di Trento (Italy).