Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2014) Volume 3, Issue 6
ATP-binding cassette (ABC) transporters are molecular motors that transport substrates through biological membranes, using the energy of ATP binding and hydrolysis to foster a series of conformational changes that follow the alternating access mechanism principles. While this general scheme of substrate transport is well accepted, details about the ATP hydrolysis mechanism itself are still a matter of debate. Decades of biochemical and structural studies have identified well-conserved key residues involved in the enzymatic reaction, but their actual role, or order in the hydrolytic process, differ according to the mechanism involved. Here, we investigate the fundamental mechanisms of ATP hydrolysis, by reporting the nucleotide-free structure of the N-terminal nucleotide-Binding Domain (NBD1) of human Multidrug Resistance Protein 1 (MRP1/ABCC1), a transporter of chemotherapeutic drugs. Comparison with the nucleotide-bound structure clearly identified movements of H827 associated with nucleotide binding and release of the active site. These results, put in the context of other structural information and mutational studies of ABC transporters, support the general-base catalysis mechanism for ATP hydrolysis for ABCC1.
<Keywords: ATP-binding cassette transporters, Nucleotide binding domain, Multidrug resistance protein 1, MRP1, ABCC1, ATP hydrolysis, General-base mechanism
Multidrug Resistance Protein 1 (MRP1), renamed ABCC1 with the current nomenclature [1], is a member of the ATP-binding cassette (ABC) superfamily of membrane transporters [2]. ABCC1 physiological role is associated with cell detoxification through the transport of organic anions, many of which being conjugated to glutathione (GSH), sulfate or glucuronate [3]. GSH itself is a substrate of ABCC1 and leukotriene C4 (LTC4), an endogenous GSH-conjugate, is the substrate with highest affinity [3-6]. ABCC1 also transports cytotoxic drugs used in cancer therapies, such as vincristine, vinblastine, or daunorubicin in co-transport with GSH [4-6]. ABCC1 thereby protects the cell against therapeutic agents and contributes to multidrug resistance.
Most ABC transporters, including ABCC1, obey the alternating access mechanism to transport substrates across biological membranes, using the energy of ATP binding and hydrolysis [7]. ATP binds at the interface between two nucleotide-binding domains (NBD), bringing the two NBDs in close proximity. This results in conformational changes of the trans-membrane domains, which will open in the outward facing conformation, permitting release of the substrate. ATP hydrolysis followed by release of inorganic phosphate and ADP resets the transporter in the inward-facing conformation, ready to bind new substrate. While this general scheme of ABC transporter function is well accepted, details of ATP hydrolysis at the fundamental level are still a matter of debate. In addition to the question whether ATP hydrolysis is sequential or alternate between the two sites (switch model) [8,9], two main mechanisms for ATP hydrolysis itself have been proposed (Figure 1): the ‘general-base catalysis’ [10-12] and the ‘substrate-assisted catalysis’ [13,14]. In the former, the catalytic glutamate residue immediately following the Walker-B motif acts as a base to deprotonate the attacking water molecule while in the later, the attacking water molecule is deprotonated by the oxygen of the γ-phosphate of ATP. This oxygen is coordinated by the histidine of the H-loop, and the glutamate following the Walker-B motif holds the histidine in a favorable position.
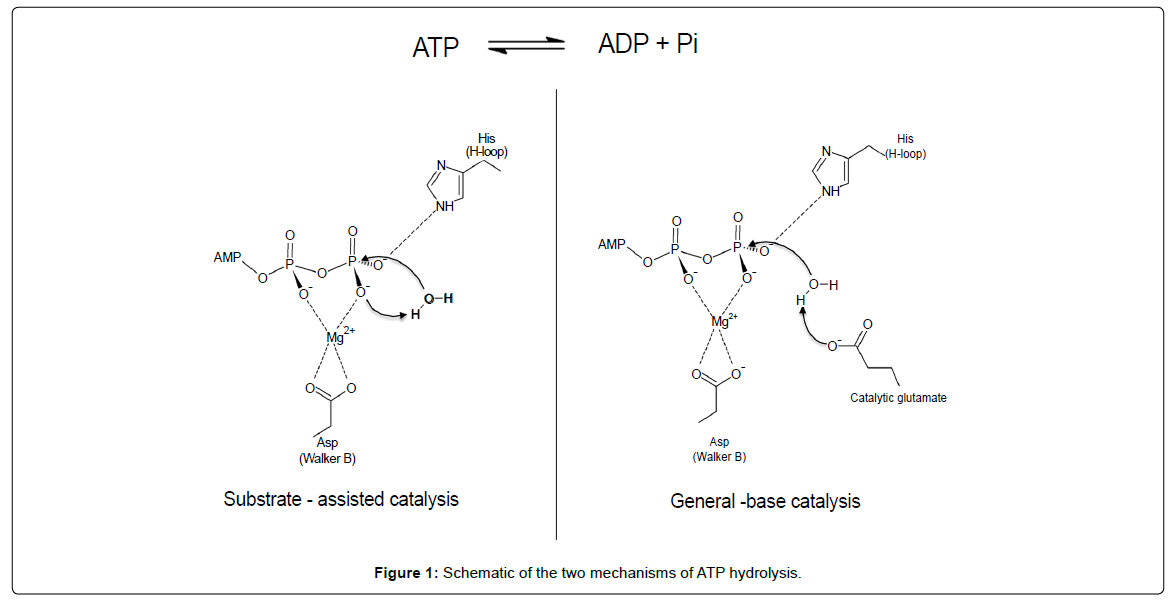
Figure 1: Schematic of the two mechanisms of ATP hydrolysis.
The two NBDs of ABCC1 are not functionally equivalent: the N-terminus NBD, NBD1, binds ATP with a higher affinity than the C-terminus NBD, NBD2, but is defective for ATP hydrolysis contrary to NBD2 [15-17]. In this paper, we investigated the reasons of this discrepancy and present here the crystal structure of nucleotide-free NBD1 showing conformational changes of key residues involved in ATP hydrolysis. These results shed new light on NBD1 structural dynamics and support the ‘general-base catalysis’ mechanism for ATP hydrolysis.
Cloning, protein expression and purification
The DNA fragment encoding residues 628 to 881 of human ABCC1-NBD1 was inserted between the BamHI and Hind III sites of the expression vector pT7-7/6His (generous gift from J.-C. Cortay, Lyon France) to insert a 6His-tag in N-terminus. E. coli BL21-DE3 (Novagen) bacteria were transformed, grown in LB media (Sigma- Aldrich) to OD600=0.7 and H6-NBD1 expression was induced with 0.4 mM IPTG at 25°C for 4 hours. After centrifugation 15 min at 5,000 xg, the cell pellet was resuspended in 50 mM Hepes pH 7.8, 0.5M NaCl, 7mM MgCl2, 1 mM EDTA. Cells were lysed using a French Press at 1000 psi after addition of antiprotease cocktail (Roche), 1mM PMSF and 30 U/ml benzonase (Sigma-Aldrich). Cell debris was removed by centrifugation 30 min at 30,000 g at 4°C. H6-NBD1 was purified by chromatography using a Nickel-loaded Hitrap Chelating HP 5 ml column (GE Healthcare). Unbound material was washed using 50mM Hepes pH 7.8, 0.5M NaCl, 5mM β-mercaptoethanol, 10% glycerol, 25 mM imidazole. H6-NBD1 was eluted on a 25 to 250 mM imidazole gradient over 6 column volumes. NBD1 was further purified by size exclusion chromatography (Superdex 200 HiLoad 16/600, GE Healthcare) equilibrated in 25 mM Hepes pH 7.8, 5mM DTT. Purity of the sample was assessed by SDS-PAGE. Freshly purified H6-NBD1 was concentrated to 15 mg/ml on 3KDa Amicon Ultra concentrators (Millipore).
Crystallization and data collection
The crystallization work benefited from the Protein Science Facility of the SFR Biosciences Lyon.) Screening was conducted using a Mosquito workstation (TTP Labtech) on commercial crystallization solutions with the sitting-drop vapour diffusion technique, against a protein solution supplemented with 5mM ATP-MgCl2. All crystallization trials were performed at 19°C and visualized on RockImager 182 (Formulatrix). Initial crystals were obtained with 10 mg/ml H6-NBD1and 5mM ATP-MgCl2 in 25% PEG 3350, 0.2M AmSO4, 0.1M BisTris pH 5.5 (JCSG+ H7 crystallization screen, Qiagen). Crystals were reproduced manually with homemade solutions and cryoprotected with the crystallization solution supplemented by 10% glycerol and flash-cooled in liquid nitrogen. Diffraction data were collected at the ESRF on beamlines ID29 and ID14-4. The NBD1 crystal belonged to space group P31, with one molecule per asymmetric unit. Data from two crystals were processed with Mosflm [18] and merged with Aimless [19]. The structure was solved by molecular replacement with PHASER [20] using the human ABCC1-NBD1 as the search model (PDB code: 2CBZ). Bias was removed with simulated annealing and the model was refined by cycles of refinement in COOT [21] and Phenix [22]. In order to obtain the nucleotide-free structure, crystals were back-soaked in 25% PEG 3350, 0.2M AmSO4, 0.1M BisTris pH 5.5. Crystals were flash-cooled in liquid nitrogen; data was collected in SOLEIL on beamline Proxima-1 from a single crystal and treated as before. The back-soaked structure displayed significant structural differences, was refined and deposited in the protein data bank under accession code 4C3Z.
To investigate the lack of ATP hydrolysis by NBD1 of ABCC1, we first solved the structure of NBD1 in complex with ATP. Our crystallization conditions were different from those described [23] and the construct used in this study contained 26 additional N-terminus residues. We observed electron density for one more residue in N-terminus and one less in C-terminus, allowing the modeling of the fragment corresponding to residues 641-871 of human ABCC1. The two structures are otherwise nearly identical. Our additional 25 N-terminus residues are not visible as they face a large solvent channel within the crystal and are disordered.
In order to obtain the nucleotide-free structure, we back-soaked the crystals in the mother liquor, containing 200 mM ammonium sulfate. The new nucleotide-free structure has well defined electron density for the whole chain allowing clear positioning of side chains and excellent modeling of the structure. ATP is no longer visible in the active site, and a sulfate ion takes the place of its alpha-phosphate (Figure 2); in addition, several other sulfate ions are seen around the protein in solvent accessible regions. The Mg2+ ion is also absent from the active site. As a result of ATP-Mg2+ departure from the P-loop, Q713 shifts 2.7 Å away from the Mg2+ coordination position, triggering movements of the Q-loop. This is in good accordance with findings that movements of the Q-loop are essential for ATP hydrolysis [24]. The loop carrying the Walker-B motif (D792) also rearranges upon ATP-Mg2+ departure. The movements of these two loops, which are critical in the ATP hydrolysis models, reveal the intrinsic plasticity of NBD1 despite the constraints generated by crystal packing. Structural rearrangements within NBDs are frequently observed in all the structures available to date [10,25-28] and reveal that NBD1 shares the same plasticity as NBDs hydrolyzing ATP. The lack of ATP hydrolysis by NBD1 is therefore not caused by a lack of internal flexibility.
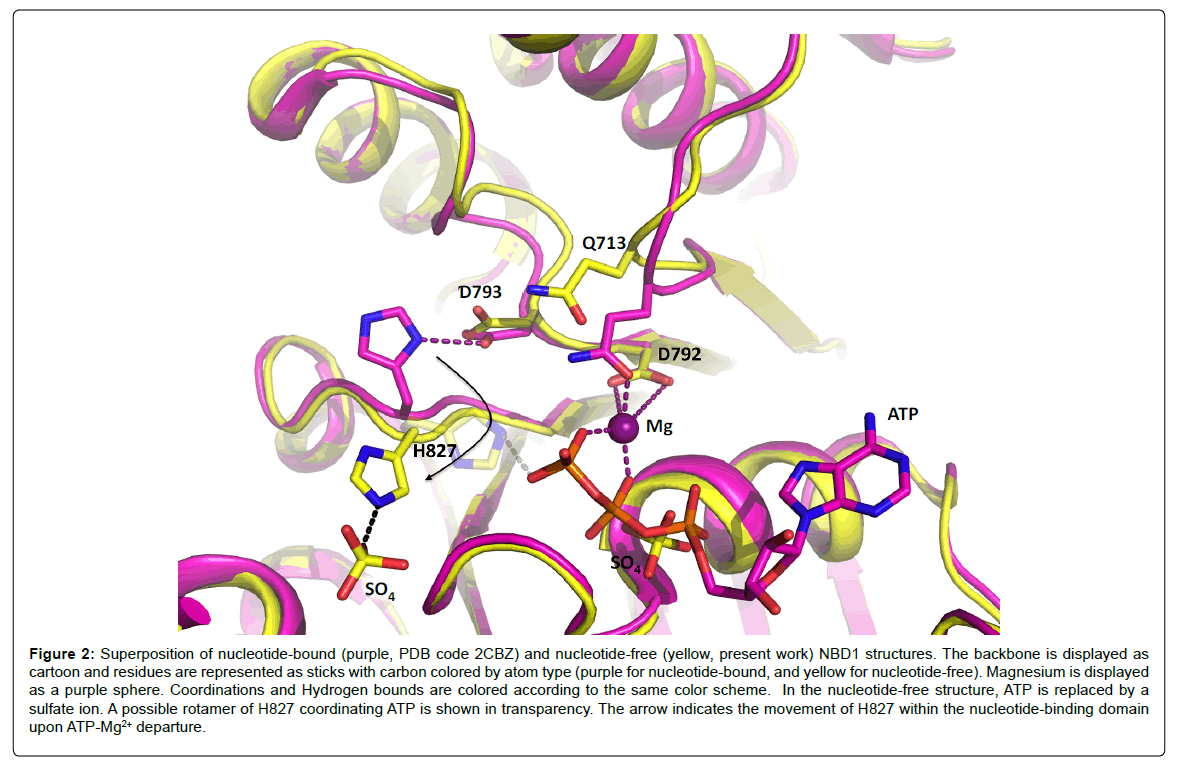
Figure 2: Superposition of nucleotide-bound (purple, PDB code 2CBZ) and nucleotide-free (yellow, present work) NBD1 structures. The backbone is displayed as cartoon and residues are represented as sticks with carbon colored by atom type (purple for nucleotide-bound, and yellow for nucleotide-free). Magnesium is displayed as a purple sphere. Coordinations and Hydrogen bounds are colored according to the same color scheme. In the nucleotide-free structure, ATP is replaced by a sulfate ion. A possible rotamer of H827 coordinating ATP is shown in transparency. The arrow indicates the movement of H827 within the nucleotide-binding domain upon ATP-Mg2+ departure.
Another significant movement observed upon ATP-Mg2+ departure is the flip of H827. In the ATP-bound structure [23], the histidine side chain was hydrogen-bonding D793, which was believed to be a strong interaction resulting in a non-productive conformation. In the present ATP-free structure, the side chain of H827 has flipped away from its original position and coordinates a sulfate ion from the solvent despite the possibility to interact with D793, still in the same position. This observation implies that the side chain of H827 is free to move and to interact with different partners in the active site. Especially, a manual change of rotamer for the side chain places the histidine Ne2 atom only 2.8 Å away from the oxygen atom of the γ-phosphate of ATP (Figure 2), which confirms that transient interactions are possible between H827 and ATP.
This placement of the histidine side chain is important for the discrimination between ATP hydrolysis models (Figure 1). Indeed, in the ‘substrate-assisted catalysis’ model, the histidine side chain interacts with the γ-phosphate of ATP, fostering deprotonation of the attacking water by the phosphate moiety itself [13]. A reasonable hypothesis is that the transient interaction with ATP inferred from the nucleotidefree structure would allow a low basal ATPase activity of NBD1, which we never detected. Ramaen et al [29,30] detected a very weak ATPase activity for NBD1 using 33P-ATP, where “a single turnover (1 ATP molecule hydrolyzed per NBD1) would take several hours”. This extremely low rate on pure NBD1 does not fit with a mobile H827 observed here, able to interact with ATP even transiently. Besides, mutation of H827 to phenylalanine does not change ATP binding to NBD1, and has no influence on drug transport [31]. The ‘substrateassisted catalysis’ model appears therefore rather unlikely for NBD1.
In the context of the full transporter, ATP hydrolysis often requires dimerization of the NBDs to occur, and sometimes in a concerted manner [8,9]. This is marked for ABC transporters of the C sub-family where some members have a degenerated NBD, which does not hydrolyze ATP. A well-described example is the transporter TmrA/B from Thermus thermophiles [32], with a degenerated NBD on TmrB, where an aspartate residue replaces the catalytic glutamate, like in the NBD1 of ABCC1. Interestingly, the mutation D>E alone does not produce the ATPase activity in TmrB, but results in ATP hydrolysis in the context of the full transporter, showing that a structural rearrangement is needed for the enzymatic activity, not only based on the mutation of a key residue. The case seems however more straightforward for ABCC1, where mutational studies on the whole transporter showed that the D793E mutation enhances NBD1 ATPase activity [15], resulting in a fully active NBD1, while the reverse mutation in NBD2 (E1455D) markedly decreased ATPase activity [15]. All the structural data available today show that the NBDs dimerize in a ‘head-to-tail’ manner putting the motif signature C in contact with the neighboring subunit and contributes to place key residues in the correct orientation for the enzymatic reaction. The ATP hydrolysis itself does require a glutamate residue following the walker B motif. It is well documented for the maltose transporter where many structures in complex with ATP catalytic intermediates support the role of the catalytic glutamate as the main base to extract the proton from the attacking water molecule [10]. In ABCC1, the presence of the glutamate is also required for catalysis [15]. In this context, our structure illustrates that the main factor for the lack of ATP hydrolysis of NBD1 is the shorter distance of the aspartate side chain compared to glutamate, preventing a fully functional active site, as H827 is able to adopt multiple conformations in the active site. Taken as a whole, these data point to the role of the H-loop histidine residue to be an important player of the active site in orienting the ATP molecule. The glutamate following the Walker-B motif is responsible for deprotonating the attacking water molecule, supporting the general-base catalysis mechanism for ATP hydrolysis.
We thank the staff of beamlines ID14 and ID29 at ESRF, and Proxima-1 at SOLEIL. This work was supported by the CNRS and University of Lyon 1 (UMR 5086), the ANR and the Ligue Nationale Contre le Cancer (équipe Labellisée Ligue 2014).