Journal of Antivirals & Antiretrovirals
Open Access
ISSN: 1948-5964
ISSN: 1948-5964
Research Article - (2010) Volume 2, Issue 1
Hendra and Nipah viruses are recently emerged zoonotic paramyxoviruses for which there is no vaccine or protective therapy available. While a number of experimental therapeutics and vaccines have recently been reported, all of these will require lengthy approval processes, limiting their usefulness in the short term. To address the urgent need for henipavirus therapeutics, a number of currently licensed pharmaceuticals have been evaluated for off label efficacy against henipavirus replication in vitro. Initially it was observed that compounds which released intracellular calcium stores induced a potent inhibition of henipaviruses replication, prompting the evaluation of known drugs with a similar effect on calcium mobilisation. Of the eight compounds randomly selected based on existing literature, seven inhibited virus replication in the micromolar range while the remaining compound also inhibited virus replication but only at millimolar concentrations. Pretreatment experiments with various calcium chelators, channel antagonists or endoplasmic reticulum release inhibitors supported a calcium mediated mechanism of action for five of these compounds. The mechanism of antiviral action for the remaining three compounds is currently unknown. Additionally, a number of other modulators of calcium flux, including calcium channel and calmodulin antagonists also exhibited potent antiviral activityin vitro providing a broad range of potential therapeutic options for the treatment of henipavirus infections. Importantly, as many of these compounds are currently licensed drugs, regulatory approval should be a much more streamlined process, with the caveat that appropriate in vivo efficacy can be demonstrated in animal models.
<Keywords: Nipah virus; Hendra virus; Henipaviruses; Antiviral; Calcium
2-APB: 2-Aminoethyl Diphenylborinate; BAPTA-AM: 1,2-bis(2-aminophenoxy)ethane-n,n,n’,n’- tetraacetic acid (acetoxymethyl ester); BHQ: 2,5-di-(t-butyl)- 1,4-hydroquinone; BSL: Biosafety Level; CA1001: (-)-(r,r)- N,N’-bis[11-(ethoxycarbonyl)undecyl]-n,n’-4,5-tetramethyl-3,6- dioxaoctanediamide; CC50: Concentration Resulting in 50% Cytotoxicity; ER: Endoplasmic Reticulum; EGTA: Ethylene Glycol-bis(2-aminoethylether)-N,N,N’N’-tetraacetic Acid; HeV: Hendra Virus; HRP: Horse Radish Peroxidase; HTS: High Throughput Screening; IC50: Concentration resulting in 50% inhibition; IP3: Inositol Triphosphate; MOI: Multiplicity Of Infection; NiV: Nipah Virus; PBS-T: Phosphate Buffered Saline Containing Tween; PKC; Protein Kinase C; PLC; Phospholipase C; PMA: Phorbol 12-myristate 13-acetate; PMCA: Plasma Membrane Calcium ATPase; SERCA: Sarco/endoplasmic Reticulum Ca2+-ATPases; SOCE: Store Operated Ca2+ Entry; TCID50: 50% Tissue Culture Infectious Dose
The genus Henipavirus is a newly formed group that includes Hendra (HeV) and Nipah (NiV) viruses (Wang et al., 2001). Both viruses are zoonotic Paramyxoviruses that are highly pathogenic with case fatality rates in humans reaching more than 75% (Halpin and Mungall, 2007). HeV was recognized as a novel paramyxovirus in 1994 following two separate outbreaks in Queensland, Australia which resulted in two human fatalities following virus transmission from infected horses (Murray et al., 1995a; Murray et al., 1995b; O’Sullivan et al., 1997). Since then HeV re-emergence has been documented numerous times (ProMed-mail, 2008) resulting in 43 equine deaths and a further human fatality in each of August 2008 (Anonymous, 2008) and August 2009 (ProMed-mail, 2009). The related Nipah virus (NiV) was identified as the cause of a lethal, febrile encephalitis in Malaysia and Singapore in 1998-1999 resulting in more than one hundred human fatalities (Chua et al., 1999). Since 2001 NiV has continued to re-emerge almost annually in Bangladesh and India causing severe respiratory distress and encephalitis, and human to human transmission has been reported (Icddr, 2003; Hsu et al., 2004; Icddr, 2004a; Icddr, 2004b; Icddr, 2005; Chadha et al., 2006) and reviewed in (Chong et al., 2008).
There are currently no vaccines or therapeutic remedies specifically indicated to either prevent or treat patients exposed to henipaviruses (reviewed in (Halpin and Mungall, 2007)). The broad spectrum antiviral compound, ribavirin, was evaluated in a limited non-randomised, open label trial during the initial NiV outbreak in Malaysia and was associated with a reduction in mortality of acute NiV encephalitis (Chong et al., 2001). In vitro studies of ribavirin have demonstrated effective inhibition of both HeV (Wright et al., 2005) and NiV (Aljofan et al., 2008). Also, a recent in vivo study showed that the 5-ethyl analogue of ribavirin, but not ribavirin, was able to prevent mortality in five of six animals in a hamster model of NiV infection (Georges- Courbot et al., 2006). During a high throughput screen using HeV pseudotyped-VSV, the anti-malaria compound, chloroquine, has been identified as an effective henipavirus antiviral in vitro (Porotto et al., 2009). However, it was recently shown that chloroquine had no antihenipavirus effect in vivo (Pallister et al., 2009), either with or without combination therapy with ribavirn (Freiberg et al., 2009).
We have also conducted a medium to high throughput screen of over 8,000 low molecular compounds against live NiV under Biosafety Level 4 (BSL4) conditions (Aljofan et al., 2008; Aljofan et al., 2009a; Aljofan et al., 2009b). In addition to nine novel compounds, this screen identified three commercially available compounds (Aljofan et al., 2009b) providing a means to rapidly evaluate their mechanism of action. One of the identified compounds, Brilliant Green, was observed to induce a rapid and sustained rise in intracellular calcium concentration. While we initially suspected this may be related to a disruption of membrane integrity due to dye intercalation, the related compound Gentian Violet, also a potent in vitro henipavirus antiviral, induced a smaller, rapid increase in intracellular calcium concentration that did not escalate over time (Aljofan et al., 2009b). Further investigation revealed compounds which modulated ion channel flux, particularly intracellular calcium flux, induced a potent inhibition of henipavirus replication in vitro. In an effort to provide clinically useful drugs, which are immediately available for henipavirus infections, we evaluated the antiviral efficacy of a number of established pharmaceuticals, previously described in the literature to modulate intracellular calcium levels. In this study we describe a number of clinically available therapeutics, effective in vitro against Hendra and Nipah virus replication. Some of these compounds may be exerting their effects via releasing intracellular calcium, providing a novel antiviral mechanism that may be applicable to a number of other related viruses.
Reagents
2,5-di-(t-butyl)-1,4-hydroquinone (BHQ); (-)-(r,r)-N,N’- bis[11-(ethoxycarbonyl)undecyl]-n,n’-4,5-tetramethyl-3,6- dioxaoctanediamide (CA1001); ryanodine and xestospongin C were purchased from Merck Scientific (Kilsyth, VIC Australia). (-)-7-octylindolactam V, (+/-)-verapamil hydrochloride, (+/ -)-propranolol hydrochloride, [arg8]-vasopressin acetate salt, 1,2- bis(2-aminophenoxy)ethane-N,N,N’N’—tetraacetic acid (acetoxymethyl ester) (BAPTA-AM), 25-hydroxy-cholecalciferol, 4-hydroxytamoxifen, bradykinin acetate, caffeine, calcium ionophore A23187, chemiluminescent peroxidase substrate-3 (CPS-3), cholecalciferol, clotrimazole, ethylene glycol-bis(2- aminoethylether)-N,N,N’N’-tetraacetic acid (EGTA), ionomycin from streptomyces conglobatus, nifedipine, phorbol 12-myristate 13-acetate (PMA), protein kinase C (PKC) inhibitor (meristoylated), ribavirin, thapsigargin and U73122 hydrate were purchased from Sigma Chemical Company (St. Louis, MO, USA). The CellTiter-Glo® cytotoxicity kit was purchased from Promega (Madison, USA). Indo-1 was purchased from Invitrogen (Carlsbad, CA, USA). RNeasy kits were purchased from Qiagen (Valencia, CA, USA). First strand synthesis Superscript III Reverse Transcriptase kits were purchased from Invitrogen (Carlsbad, CA, USA). Jumpstart SYBRGreen real time PCR reagent was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Virus and cells
African Green Monkey Kidney (Vero) cells were grown in Minimal Essential Medium containing Earle’s salts (EMEM), antibiotics (100 U penicillin, 100 μg/ml streptomycin and 500μg/ml Fungizone) and 10% fetal calf serum (FCS), designated EMEM-10. NiV was isolated in Vero cells from the brain of a human fatally infected in the 1998-99 Malaysian outbreak and was passaged three times in Vero cells then double plaque purified and passaged a further three times in Vero cells as previously described (Shiell et al., 2003). HeV was isolated in Vero cells from the lung of a horse infected in the Brisbane outbreak in October 1994 and was passaged five times in Vero cells followed by triple plaque purification and a further five passages in Vero cells as previously described (Hyatt et al., 1996). HeV and NiV stock titer were adjusted to 1×106 TCID50/ml. All work with live virus was carried out under Biosafety Level 4 (BSL 4) conditions at CSIRO Australian Animal Health Laboratory (AAHL).
Henipavirus infection of Vero cells and antiviral screening
Vero cells were seeded at a density of (2x104) into individual wells of 96-well microtitre plates and incubated at 37°C overnight in 100 μl EMEM-10. Prior to NiV inoculation, medium was discarded and 100 μl of 20 μM of different test compounds were added to each well in triplicate. Under BSL4 conditions, 1,000 TCID50 of virus in EMEM-10 were added to each well of Vero cells in volumes of 100 μl (final compound concentration of 10 μM). After an overnight incubation at 37°C, the culture medium was discarded; plates were immersed in ice-cold absolute methanol, enclosed in heat sealed plastic bags and the bags surface sterilized with Lysol for removal from the BSL4 laboratory. Methanol-fixed plates were air dried at room temperature for a minimum of 30 minutes prior to immunolabeling. In pretreatment experiments cells were treated for 60 minutes with one of the following compounds: 10 μM BAPTA, 10 μM EGTA, 5 mM calcium chloride, 10 μM xestospongin C, 1 μM ryanidine or 10 μM verapamil. Medium was then removed and cells were washed with fresh media. Test compound was then applied at half-log concentrations, immediately followed by the addition of virus (1,000 TCID50 in EMEM-10). Viral infection was quantified as described above.
HTS immunolabeling assay
Virus replication was assessed as previously described (Aljofan et al., 2008). Briefly, plates were washed 3 times with phosphate buffered saline containing 0.05% Tween-20 (PBS-T). Plates then were protein blocked with 100 μl of 2% skim milk in PBS-T and incubated at 37°C for 30 minutes. After blocking, plates were washed 3 times with PBS-T, followed by incubation with 100 μl anti-NiV-N antibody (prepared by AAHL Bioreagents Development Group) diluted 1:1,000 in PBS-T containing 2% skim milk for 30 minutes at 37°C and then washed 3 times with PBS-T. Plates were incubated with 1% H2O2 (Sigma) for 15 minutes at room temperature then washed with PBS-T 3 times. Aliquots of 100 μl of goat anti-rabbit HRP conjugated antibody (Sigma) diluted 1:2,000 in PBS-T containing 2% skim milk, were added to each well and plates incubated at 37°C for 30 minutes. Plates were washed 3 times with PBS-T and 100 μl aliquots of Chemiluminescent Peroxidase Substrate-3 (CPS-3, Sigma) diluted 1:10 in chemiluminescent assay buffer (20 mM Tris-HCl, 1 mM MgCl2, pH 9.6) were added to all wells. Plates were incubated at room temperature for approximately 15 minutes, and then read using a Luminoskan Ascent luminometer (Thermo Fisher Scientific, Waltham, USA) using 100 millisecond integration per well.
Antiviral cytotoxicity assay
Compounds were evaluated for cytotoxicity using the CellTiter- Glo cytotoxicity kit (Promega) according to the manufacturer’s instructions. The CellTiter-Glo Luminescent Cell Viability Assay is a homogeneous method of determining the number of viable cells in culture based on quantitation of the ATP present, which signals the presence of metabolically active cells. Briefly, Vero cell monolayers were incubated with 200 μl of half-log dilutions in EMEM-10 (20 μM-63 nM) of each compound (n=3) overnight at 37°C. Medium was removed and 100 μl of CellTiter- Glo Reagent, diluted 1:5 with chemiluminescent assay buffer, was added to each well, mixed well to lyse cells, equilibrated to room temperature for 10 minutes, and then read using a luminometer as described above. Non-linear regression analysis performed using GraphPad Prism software to determine the concentration resulting in 50% cytotoxicity (CC50).
Determination of cytokines gene expression
Vero cells of approximately 1 x 105 densities, were seeded in 48 well plates, and treated with compounds at various concentrations (10 μM, 5 μM, and 1.25 μM) for 24 hours. RNA was extracted using Qiagen RNeasy kits. RNA from each sample (1μg) was treated with DNase I (Invitrogen) for 15 minutes, reverse transcribed with 0.5 μg oligo dT(12-18) primers (Invitrogen) using Superscript II RT (Invitrogen), and diluted 1:5 before 2 μl of each diluted sample was subject to real-time PCR in triplicate using Jumpstart SYBRGreen kits (Sigma) on an ABI 7900HT Realtime PCR system (Applied Biosystems). Real-time PCR primers specific for TNF-α and IL-8 were obtained from SuperArrayBiosciences (Applied Biosystems) and were used at a concentration of 200 nM for each reaction. Fold change in mRNA transcripts over mock levels were determined using the delta-delta-Ct method.
Effect of intracellular calcium releasing compounds on henipavirus replication
To examine the effect of intracellular calcium release on viral infection the application of thapsigargin, a potent releaser of inositol triphospate (IP3) dependent ER calcium stores (Treiman et al., 1998; Thomas et al., 1999) induced highly effective inhibition of henipavirus replication in vitro (Figure 1) as did the calcium ionophore, A23187, which makes permeable to calcium both the plasma membrane (allowing influx of extracellular calcium) and to the ER membrane facilitating calcium efflux into the cytosol. The addition of other calcium ionophores also resulted in the inhibition of henipavirus replication, but their efficacy varied by several orders of magnitude (A23187 >> ionomycin > CA1001), perhaps reflecting differences in their specificity for calcium transport (Table 1). Similarly, BHQ, another releaser of calcium from intracellular stores via IP3 stores also inhibited virus replication, but was over 100-fold less potent than thapsigargin. We also observed that the IP3 mediated calcium release antagonists, 2-APB and xestospongin C, inhibited virus replication at concentrations similar to BHQ (xestospongin C) or greater (2-APB) as did an inhibitor of ryanodine-mediated calcium release from the ER (dantrolene) in addition to ryanodine itself (Table 1).
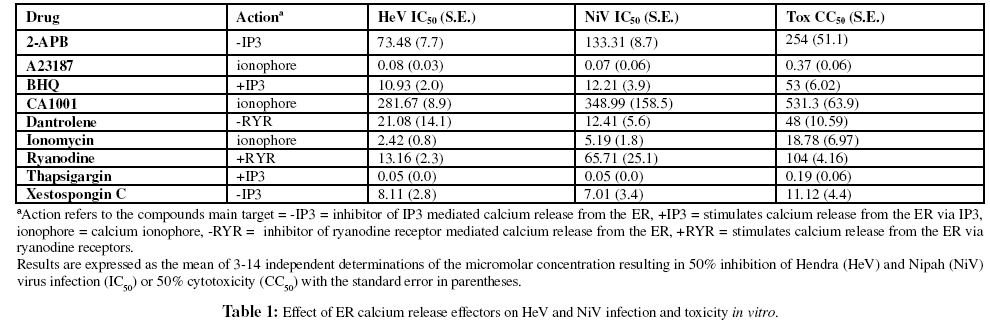

Figure 1: Effect of A23187 and thapsigargin addition on henipavirus replication in vitro. Inhibition of HeV and NiV replication as measured by viral nucleocapsid protein expression determined by immunoassay. Each compound was incubated with cells for 60 minutes, media was then removed and virus inoculum (1,000 TCID50/ml NiV) added and incubated for 60 minutes, then inoculum was removed and replaced with EMEM-10 and cells were incubated overnight. Viral protein was immunoassayed as previously described (Aljofan et al., 2008). Values are expressed as the mean +/- S.E. (n=3). In parallel, cell cytotoxicity assays were performed to determine the relative toxicity of each compound as described in the materials and methods. Non-linear regression analysis was performed using GraphPad Prism.
Modulation of antiviral efficacy by calcium chelation
The calcium ionophore, A23187, mediated viral replication inhibition of HeV, which was partially reduced by either the removal of extracellular calcium through the addition of EGTA or the addition of exogenous calcium (Figure 2) suggesting at least some of their effects may be due to the flux of calcium either into or out of the cell. EGTA had no effect on thapsigargin efficacy supporting the hypothesis that its antiviral effect is mediated through release of intracellular calcium. In contrast, A23187 mediated inhibition of NiV replication was not significantly modulated by calcium chelation. For both A23187 and thapsigargin, chelation of intracellular calcium using BAPTAAM resulted in approximately 30-50% reduction of antiviral efficacy of thapsigargin and A23187, respectively against HeV, highlighting the importance of available cytosolic calcium for antiviral efficacy with these agents. Individual addition of neither BAPTA, EGTA, nor exogenously added calcium exhibited considerable inhibition of virus replication (data not shown).

Figure 2: Effect of modulators of intracellular calcium release on the efficacy of A23187 and thapsigargin inhibition of henipavirus replication. Vero cells were pretreated for 60 minutes with 10 μM BAPTA, 10 μM EGTA, 5 mM calcium chloride, 10 μM xestospongin C, 1 μM ryanidine or 10 μM verapamil prior to the addition of 10 nM A23187 or 100 nM thapsigargin. Cells were infected, incubated and immunoassayed as above. Values are expressed as the mean +/- S.E. (n=3).
Blockade of intracellular calcium release from either IP3 dependant stores (xestospongin C) or ryanodine sensitive stores (ryanodine) partially reversed the inhibition due to thapsigargin and A23187 (Figure 2). Additionally, pretreatment with a calcium channel antagonist (verapamil) significantly reduced virus replication, further supporting the possibility that these antiviral effects are partially mediated by the flux of calcium across the plasma membrane. While thapsigargin exerts its effect by inhibiting sarco/endoplasmic reticulum Ca2+-ATPases (SERCA) (Paula et al., 2004), thus depleting the ER calcium stores and increasing the cytosolic calcium levels, virtually all of this, mobilised cytosolic calcium, is immediately extruded from the cell via plasma membrane calcium ATPase (PMCA) (Hong et al., 1999).
This potentially provides a common pathway with A23187 involving calcium efflux. Given the relative modulation by EGTA of the antiviral effects exerted by these compounds, it is possible that A23187 relies on calcium influx as well, while thapsigargin exerts its effects via calcium efflux.
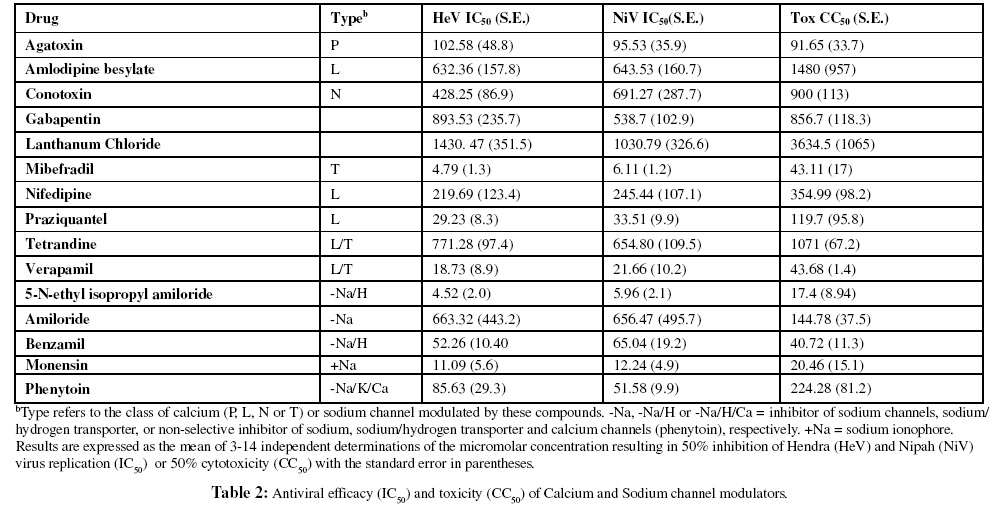
Effect of calcium and sodium channel antagonists
To further evaluate the direct role of ion channel blockade on viral infectivity, ten calcium channel antagonists, four sodium channel antagonists and one sodium ionophore were evaluated for direct effect on viral infectivity. Of the calcium channel antagonists, only mibefradil, verapamil and praziquantel afforded considerable reduction in viral infectivity in the low micromolar range (Table 2). Interestingly, despite the direct inhibitory effects of verapamil, it significantly reversed the inhibition associated with all other calcium antagonists (data not shown). The most significant effect of sodium channel antagonists was evident with 5-N-ethyl isopropyl amiloride (EIPA) followed by benzamil. Both compounds are inhibitors of the Na/H antiport (Abrahamse et al., 1994) suggesting a possible role for pH modulation of virus replication. Phenytoin and amiloride were less effective at inhibiting either virus while the sodium ionophore, monensin, was effective in the low micromolar range (Table 2). Monensin has previously been shown to inhibit HeV F protein processing (Pager et al., 2004) and this was attributed to the inhibition of vesicular transport between the medial and trans- Golgi cisternae (Griffiths et al., 1983).
Further investigation of additional calcium modulators revealed that the calmodulin antagonist, phenoxybenzamine, also resulted in considerable inhibition of viral replication suggesting the level of free calcium intracellularly is an important factor in viral infection (Table 3). The fungal metabolite gliotoxin, reported to suppress rises in intracellular calcium concentration via store operated Ca2+ entry (SOCE) in mast cells (Niide et al., 2006), also produced potent inhibition of viral infectivity, as we described earlier (Aljofan et al., 2009b), as did cyclosporine, which, is also an activator of SERCA, which is reported to result in removal of cytosolic calcium (Hadri et al., 2006).
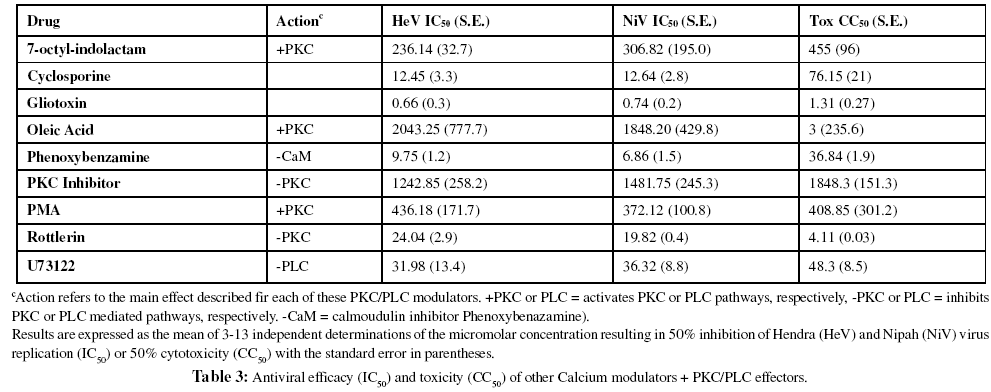
Effect of PKC activators and inhibitors on henipavirus replication
As calcium regulation is inherently linked to protein kinase C (PKC) pathways, we also examined a range of specific PKC activators and inhibitors for the potential henipavirus antiviral efficacy. Both rottlerin (PKC inhibitor) and U73122 (phospholipase C (PLC) inhibitor) produced moderate inhibition of viral replication but the effects of other PKC activators or inhibitors was quite variable. Interestingly PKC activators (PMA and 7- octyl-indolactam) inhibited virus replication at high concentrations (>10 μM), presumably through acute toxicity (Figure 3), but actually enhanced replication in the nanomolar range. Note the absence of effect of the PKC inhibitor in the nanomolar range but efficacy (without toxicity) at high micromolar concentration.

Figure 3: Inhibition of henipavirus replication by modulators of PKC activity.
Vero cells were pretreated for 60 minutes with log dilutions of PKC inhibitor, PMA or 7-octylindolam (50 μM max). Cells were then infected, incubated and assayed as above. Values are expressed as the mean +/- S.E. (n=3). In parallel, cell cytotoxicity assays were performed to determine the relative toxicity of each compound as described in the materials and methods.
Effect of therapeutically licensed drugs on henipaviruses
The potent antiviral actions observed with both A23187 and thapsigargin, and to a lesser extent by gliotoxin and phenoxybenzamine, possibly attributable to the modulation of free calcium in the cytosol has prompted us to evaluate clinically available compounds, which are reported to act as modulators of cytosolic calcium, as potential antivirals (Table 4). Of the eight compounds randomly selected based on reports in the literature of modulation of intracellular calcium, seven compounds inhibited henipavirus replication in the low micromolar range while caffeine also inhibited virus infectivity, but only in the millimolar range (Table 5). Of these, only five showed a strong dependance on intracellular calcium for their inhibition effects, determined through a series of pretreatment experiments with various calcium chelators, channel antagonists or ER release inhibitors (Table 5). Importantly, antiviral efficacy was enhanced by pretreatment of cells with thapsigargin with four of these compounds. In general, supplementation of the media with calcium chloride 10 μM prior to and during drug treatment produced similar levels of efficacy inhibition as did the removal of calcium via EDTA. This suggests calcium flux modulation by these compounds may involve the release of calcium from intracellular stores, in addition to the influx of calcium from the extracellular space. As either process may trigger the other, the order of this sequence may vary for each compound. Interestingly, the least effective of these compounds, caffeine, is reported to release ryanodine sensitive calcium stores in skeletal muscle cells (Chen et al., 1996). As supported by the lack of modulation by thapsigargin in the current study (Table 5). The five compounds with greater efficacy in this study have been reported to release IP3 sensitive calcium (Reilly et al., 1998; Sitrin et al., 1999; Wang et al., 2001; Sergeev et al., 2005) suggesting there may be an IP3 dependent portion to the antiviral effects attributable to many of these compounds. While clotrimazole exhibited a weak dependance on intracellular calcium, the effects of 4-hydroxy tamoxifen and propranolol were insensitive to changes in calcium availability, suggesting their antiviral efficacy is likely to be via an unrelated mechanism.
Determination of cytokine gene expression following antiviral treatment
While these studies provide support for the hypothesis that the antiviral efficacy of a number of these compounds is associated with the release of intracellular calcium, this release alone may have profound effects on a broad range of cellular processes including general cellular inflammatory processes. In an effort to evaluate the possibility of changes in cytokine production as an indicator of general inflammation, we evaluated the expression of two inflammatory cytokines, IL-8 and TNF- a, in response to a number of drug treatments including A23187, thapsigargin, 4-hydroxytamoxifen (as a representative of the ER calcium releasing drugs), calcium channel antagonists and PKC modulators (Figure 4). As expected our control compound, PMA stimulated an almost 60-fold increase in production of both these cytokines, while several compounds induced greater than 20-fold increase in IL-8 expression, without accompanying increase in TNF-a production. Interestingly, verapamil and mibefradil produced a much greater stimulation of IL-8 expression than did another calcium antagonist, nifedipine, partially reflective of their relative antiviral potency. Neither 4-hydroxytamoxifen nor thapsigargin induced obvious changes in cytokine expression suggesting their antiviral effects may be distinct from general cellular anti-viral responses via inflammation.

Figure 4: Expression of IL-8 and TNF- in response to antiviral addition to vero cells. Vero cells were treated overnight with 10 μM 4-hydroxytamoxifen, 10 μM EIPA, 1 μM A23187, 10 μM cyclosporin, 1 μM gliotoxin, 10 μM mibefradil, 1μM neomycin, 100 μM nifedipine, 100 nM PKC inhibitor, 1 μM PMA, 10 μM ribavirin, 100 nM thapsigargin or 100 μM verapamil. Media was then removed and RNA extracted using a Qiagen RNEasy kit as per the manufacturer’s instruction. IL-8 and TNF- expression was quantified following RNA transcription and real time PCR employing SYBR green as described in the materials and methods. Fold change in mRNA transcripts over mock levels were determined using the delta-Ct method. Results are expressed as the mean +/- S.E. (n=3 replicate determinations of each of 3 biological replicates).
The continual re-emergence of both HeV and NiV in Australia and Bangladesh, respectively, suggests that we can neither predict an outbreak, nor can we estimate their catastrophic effect. The relatively small number of animal and human cases globally makes vaccine production uneconomical, despite recent reports of viable candidate vaccine studies in three different animal models (Guillaume et al., 2004; Mungall et al., 2006; Weingartl et al., 2006). Therefore, there is an imperative need to develop an effective antiviral therapy capable of treating henipavirus infections. Currently, the only antiviral evaluated as a post-exposure therapeutic has been the broad spectrum compound ribavirin (Chong et al., 2001), a drug associated with a range of side effects primarily related to haemolytic anaemia (De Franceschi et al., 2000). Despite the lack of efficacy seen with chloroquine administration in ferrets and hamsters, there is currently minimal correlative evidence for antiviral efficacy between these models and humans. Importantly, animal model failures of safe and approved compounds in humans should not exclude the possibility of human administration in outbreak situations.
A number of therapeutic approaches have been evaluated and these included; fusion inhibitory peptides (Bossart et al., 2002; Bossart et al., 2005; Porotto et al., 2006), receptor blockade using a soluble virus glycoprotein (Bossart et al., 2005), induction of IFN via injection of poly(I)-poly(C12U) (Georges- Courbot et al., 2006), RNA interference (Mungall et al., 2008), the endogenous lectin galectin-1 (Levroney et al., 2005), mannose binding lectin (B. Mungall personal communication) and quinolone derivatives (Niedermeier et al., 2009) and humanised monoclonals (Bossart et al., 2009). Importantly, a number of laboratories have begun screening large libraries of novel compounds in the quest to discover novel antiviral candidates. It has been recently demonstrated in vitro anti-henipaviruses activity with chloroquine (Porotto et al., 2009), a safe and widely used therapeutic used in humans to treat malaria. The fact that chloroquine is already in clinical use would help to bypass many of the barriers normally associated with drug development. However, recent studies in ferrets and hamsters, reported chloroquine as ineffective in preventing a lethal NiV infection (Pallister et al., 2009; Freiberg et al., 2009). While chloroquine was identified during a high throughput screen of over 23,000 compounds using a HeV pseudotype assay, our laboratory has also partially screened an unrelated library directly against live NiV at BSL4 (Aljofan et al., 2008) resulting in a small number of potential antiviral candidates (Aljofan et al., 2009a; Aljofan et al., 2009b). While these may be important drugs for the future, the urgent need for anti-henipavirus treatments and the immediate advantages of existing drugs encouraged us to evaluate known drugs as potential antivirals.
As a result of preliminary investigations into the mechanism of action of these novel antiviral candidates, in addition to an acknowledgment of the important role that calcium plays in many physiological disease states, particularly viral infection, we initially investigated intracellular calcium modulators as potential anti-henipavirus therapeutics. Calcium is one of the most universal and versatile signaling molecules and is involved in almost every aspect of cellular processes. There is a growing body of evidence supporting the importance of calcium in the lifecycle of many viruses although this has not previously been reported for henipaviruses. In contrast, Pager et al. (2004) demonstrated that the proteolytic activation of Hendra virus F protein had a lower calcium requirement compared to that for furin-mediated activation and cleavage was not significantly affected by decreases in cellular calcium levels. The authors showed that depletion of extracellular calcium with EGTA had no effect on F protein processing while A23187 treatment produced only a minor effect at the highest concentrations (Pager et al., 2004), suggesting the inhibition observed in the current study is not related to perturbations in F protein processing.
In the current study, the partial reversal of the potent inhibition of HeV and NiV replication by A23187 and thapsigargin by the intracellular calcium chelator, BAPTA, and less so by the extracellular calcium chelator, EDTA, supports the notion that this inhibition is at least partially mediated by intracellular calcium levels. Interestingly, blockade of intracellular calcium release from either IP3 dependant stores (xestospongin C) or ryanodine sensitive stores (ryanodine) only partially reversed this inhibition while the pretreatment with a calcium channel antagonist (verapamil) significantly reduced virus inhibition suggesting much of this effect is mediated by the inward flux of calcium. Alternately, the release of calcium from intracellular stores via store operated channels (Armesilla et al., 2004), sensitive to verapamil (and potentially other calcium channel antagonists) could also explain this phenomenon.
To examine if the antiviral effects of these compounds may be a result of general cellular inflammatory processes, we evaluated the expression of two cytokines with key roles in these processes. While A23187 treatment was associated with an increase in the expression of IL-8, and to a lesser extent, TNF-α, thapsigargin did not induce a change in the expression of these inflammatory mediators, suggesting a more specific mode of action.
While these observations on their own do not definitively prove the involvement of intracellular calcium or its regulation in the mechanism of antiviral efficacy, the overwhelming body of evidence compelled us to evaluate a number of currently licensed drugs reported to effect calcium metabolism. Some of these compounds, however, were shown to have calcium dependent antiviral activity suggesting a common mode of action. Unfortunately, it is beyond the scope of this article to provide a complete review of the in vitro actions of each of these compounds, but this will be discussed as ongoing research into the mechanisms of each reported compound. The data presented have, therefore, been selected to focus attention on those compounds that might provide valuable contributions towards finding a safe and effective anti-henipavirus antiviral treatments.
One possible theory that might provide a general explanation of the antiviral mechanisms of these compounds relies on the fact that the plasma membrane provides the first barrier against virus infection. Consequently, different calcium channels (voltage-operated, receptor-operated, or store-operated) are responsible for extracellular calcium flux into the cytosol (Zhou et al., 2009). During virus infection, these plasma membrane Ca2+-signaling components often become the immediate target of attack. Therefore, by releasing the intracellular calcium into the cytosol and increasing the cytosolic calcium concentration may disable these Ca2+ channels thus making them less susceptible to viral infection. This theory might also explain the antiviral activity of the Ca2+ channel antagonists, that is by disabling the channels via drug interaction, they become less susceptible to viral infection. Hence, restriction of calcium flux may provide the cell as a whole with possible partial protection against viral infection.
The periodic re-emergence of NiV in Bangladesh and HeV in Australia, the high fatality rates associated with these infections and the recent reports of NiV human-to-human transmission ensures international attention is focused on the development of therapeutics for these two viruses. While experimental therapeutics may provide the treatments of the future, ultimately, the identification of currently available therapeutic compounds for off-label use may provide a more rapid solution to treat henipavirus infections. As we have seen with chloroquine, appropriate validation in an animal model may or may not support these antiviral effects in vivo. Therefore, further studies should incorporate a wider range of evaluation conditions including additional types of cell lines and additional virus strains to provide greater confidence in these observations prior to in vivo or clinical studies. However, the current study does provide an important first step in the identification of potential low-cost, readily available antivirals for these currently untreatable pathogens.
Conflict of Interest
The authors declare that they have no conflicting interests.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy And Infectious Diseases or the National Institutes of Health.
Funding source
This study was supported in part by a NIH, National Institute of Allergy And Infectious Diseases (NIAID) R21AI072396 developmental award to BM.
BM is supported by a CSIRO Julius Award, MA is supported by a CSIRO, OCE PhD scholarship and ML is supported by Oak Ridge Institute for Science and Education.