Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Review Article - (2014) Volume 3, Issue 6
The discovery of cis-platin in the treatment of cancer there has been a considerable exploration on the antitumoral activity of other transition metal complexes. One of the main problems about the application of transition metal complexes for chemotherapy is their potential toxicity. Recently the attention has been focused on titanium based complexes, which could have significant potential effect against solid tumor. The advantage of Ti (IV) complexes is their relative biological compatibility, which mostly leads to mild and revisable side effects. However, the hydrolytic instability of known Ti(IV) complexes and formation of different species upon water addition makes their therapeutic application problematic, and raises a strong interest in the development of relatively stable Ti(IV) complexes with well defined hydrolytic behavior that demonstrate appreciable cytotoxic activity. Strong ligand binding is also of interest to avoid complete ligand stripping by transferrin, so that the ligand may be used as a target for structure–activity relationship investigations. Titanocene dichloride (Cp2TiCl2) shows an average antiproliferative activity in vitro and promising result in vivo. Recent work has been performed in developing therapeutic analogues of Cp2TiCl2 by varying the central metal, the labile ligands (Cl) and the biscyclopentadienyl moiety. In particular, small changes to the Cp ligand can strongly affect the hydrolytic stability and water solubility properties of the metallocenes and have an impact on the cytotoxic activity. In this review we want summarize the importance of different organo-mettalic compounds in cancer therapy with focus on possible structure modification.
<Keywords: Cis-platin, Ligands, Metallo-enzymes, NMR studies
Biomedical inorganic chemistry, known as elemental medicine, is an important new area of chemistry. It really offers the potential for the design of novel therapeutic and diagnostic agents and hence for the treatment and understanding of diseases which are now intractable [1-3]. Inorganic elements has a crucial roles in biological and biomedical processes, and it is evident that many organic compounds used in medicine do not have a purely organic mode of action; some of them are activated or biotransformed by metal ions including metallo-enzymes [4], others have a direct or indirect effect on metal ion metabolism. Medicinal inorganic chemistry offers real possibilities to pharmaceutical industries, which traditionally have been dominated by organic chemistry alone, for the discovery of novel drugs with new mechanisms of action. This field has been stimulated by the success of cisplatin, the worlds best selling anticancer drug, and platinum complexes with reduced toxicity, oral activity, and activity against resistant tumors are currently on clinical trial (Figure 1). The organo-metallic complex titanocene dichloride has also been injected into patients, and Ruthenium III (RuIII) complexes show promising anti-metastatic activity. There has been recent progress in understanding the coordination chemistry and biochemistry of older metallo-drugs such as gold antiarthritic and bismuth antiulcer drugs, and further work might lead to their more effective use. Current areas with exciting clinical potential include vanadium insulin mimics, manganese superoxide dismutase mimics, lanthanide-based photoensitizers, ruthenium nitric oxide scavengers, and metal-targeted organic agents. The increasing knowledge of organo-metallic chemistry will provide scope for the design of new compounds (both inorganic and organic) in cancer therapy.
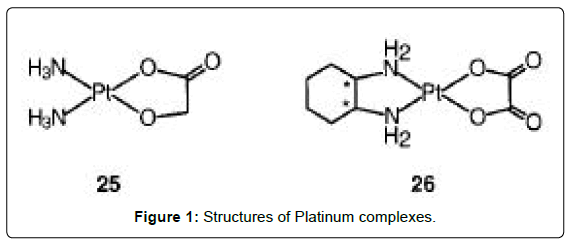
Figure 1: Structures of Platinum complexes.
Clinical Platinum Complexes
Platinum complexes are the most widely used drugs for the treatment of cancer. Four injectable PtII compounds have been approved for clinical use and several other cis-diam(m)ine complexes are on clinical trials, including an oral PtIV complex. Cisplatin (Figure 1- complex 23) is one of the most widely used anticancer drugs, together with the second generation drug carboplatin [Pt(NH3)2(CBDCA-O,O')] (Figure 1- 24). The glycolato nedaplatin, 254-S-(Figure 1- complex 25), and oxaliplatin, l-OHP, which contains R,R-1,2- diaminocyclohexane, DACH (Figure 1- complex 26) have been approved for clinical use in Japan and France, respectively. These drugs are particularly effective in combination chemotherapy for treatment of advanced lung, colorectal, and ovarian cancers [5-7]. The sterically-hindered complex ZD0473 (Figure 2- complex 27) is active (by injection and oral administration) against an acquired cisplatin- resistant subline of a human ovarian carcinoma xenograph, and entered clinical trials in 1997 [8,9]. It is less reactive than cisplatin, for example inducing DNA interstrand crosslinks in cells and binding to plasma proteins more slowly. The hydrolysis rate of ZD0473 is two to three times slower those of cisplatin. The orally active complex JM216 (Figure 2- complex 28) is reported to have superior in vitro and in vivo activity compared to cisplatin against human cervical, small cell lung, and ovarian carcinoma cell lines [10]. When JM216 is incubated with human plasma it is converted into at least six biotransformation products, which include monoand dihydroxo PtIV complexes and the dichloroplatinum(II) complex cis-[PtCl2(NH3)(cyclohexylamine)] as the major metabolite [11,12]. Complex Lobaplatin (D-19466) was introduced into clinical trials [13]. The trinuclear complex BBR3464 (Figure 3- complex 30), in which the central Pt unit is capable only of hydrogen-bonding interactions with DNA is reported to be up to 100-fold more potent than cisplatin against human tumor cell lines resistant to cisplatin [14,15]. The overall charge enhances DNA affinity, characterized by long-range interstrand cross-linking up to six base pairs apart with significant unwinding and efficient, irreversible conversion of B- to Z-DNA. The adducts terminate DNA synthesis in vitro. The cytotoxicity of the complex is insensitive to the p53 status of cisplatin resistant cells (effective against tumors carrying a mutant p53). cis- Bis(nonadecanoato)(trans-R,R-1,2-diaminocyclohexane)- platinum(II) (N-DDP) is a liposome-incorporated lipophilic cisplatin analogue that has shown promising in vivo activity against tumors resistant to cisplatin and liver metastases [16].
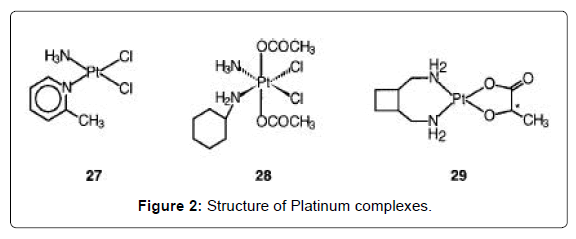
Figure 2: Structure of Platinum complexes.
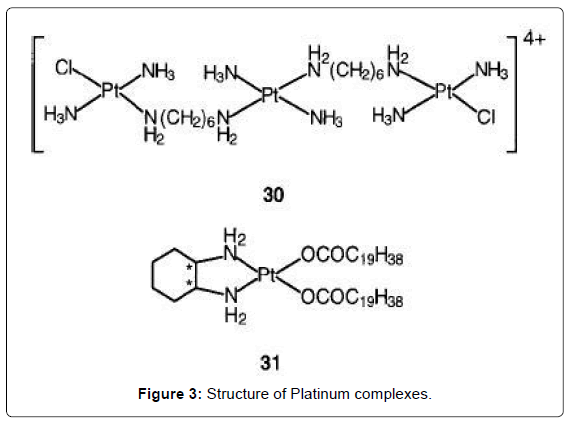
Figure 3: Structure of Platinum complexes.
Palladium Complexes as Alternative Potential Antitumor Agents
The hydrolysis of leaving ligands in Palladium (Pd) complexes is too rapid, 105 times faster than their corresponding platinum analogues. The notable analogy between the coordination chemistry of Pt(II) and Pd(II) compounds has advocated studies of Pd(II) complexs as antitumor drugs [17,18]. A key factor that might explain is most useful come from the ligand-exchange kinetics. They dissociate readily in solution leading very reactive species that are unable to reach their pharmacological targets. In addition, some of them undergo an inactive trans-conformation. This considerably higher activity of palladium complexes implies that if an antitumor palladium drug is to be developed, it must somehow be stabilized by a strongly coordinated nitrogen ligand and a suitable leaving group. If this group is reasonably non labile, the drug can maintain its structural integrity in vivo long enough. Various simple Pd(II) compounds with interesting biological properties have been previously reported such as [cis-(NH3)2PdCl2] (Figure 4- complex 1), [trans-(NH3)2PdCl2] (Figure 4- complex 2). Recent advances in this field have also focused on Pd(II) compounds bearing bidendate ligands as a way to prevent any possible cis-trans isomerism [19-21] .
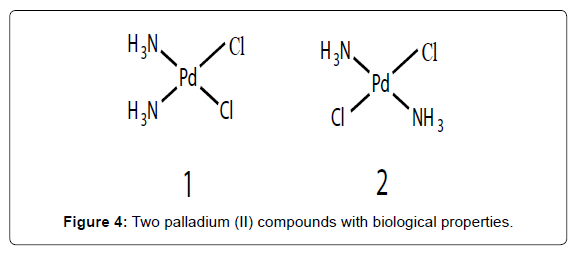
Figure 4: Two palladium (II) compounds with biological properties.
Trans-Palladium (II) Complexes Containing Bulky Monodentate Ligands
Several trans-Pd complexes with promising activity against tumor cells have been synthesized. In general, the strategies that have been applied to design these agents were on the window of reactivity usually employed for the potential platinum antitumor drugs. A comparative study on antitumor activity was carried out between the dihalide Pd(II) complexes of monoethyl-2-quinolmethylphosphonate (2-Hmqmp) and diethyl-2- quinolmethylphosphonate (2-dmqmp) [22] . The diester ligand has two potential donors, the N from quinoline and the O from phosphoryl giving the complex [trans-(2-dqmp)2PdCl2] (Figure 5- complex 1). The complexes of the diester 2-dmqmp were found to be more active than those of the monoester-based ligand (2-Hmqmp). This may partly be ascribed to the greater leaving activity of the halogen ligands in the complex bearing the 2-dmqmp ligand and to their greater lipophilicity or solubility. Fulrani et al., reported about the synthesis and cytotoxicity evaluation of some trans-[(L)2Pd(X)2] complexes (Figure 5- complex 2). Other palladium complexes based on 2-Mercaptopyridines (MP) were also prepared. The [(MP)3Pd(Br)]Br (Figure 5- complex 3) is of potential therapeutic use since it has lower IC50 values on LoVo cell lines than cisplatin and around the same as its Pt(II) analogue [23]. Pd(II) complexes containing alkyl phosphonates derived from aniline and quinoline were reported. Most of the aniline compounds (Figure 5- complex 4) showed cyctotoxicity in vitro against animal and human tumor cell lines. The palladium complex which contains the bulky nitrogen ligand harmine (7-methoxy-1-methyl- 911-pyrido [3,4-b]indole, trans-[Pd(harmine) (DMSO)Cl2] (Figure 5- complex 5) exhibits a greater cytotoxic activity against L1210 and K562 cell lines than cisplatin [24]. More recently, it has been reported the synthesis and molecular structure of a new enantiometrically pure, chiral transpalladium( II) complex, trans-[Pd{(R)-(+)- bornyl-amine}2Cl2] (Figure 6) that bears the bulky amine ligand R-(+)-bornyl-amine. The complex showed similar antitumor activity against HeLa cells when compared with the activity of the standard references, cisplatin, carboplatin and oxaliplatin [25-110].
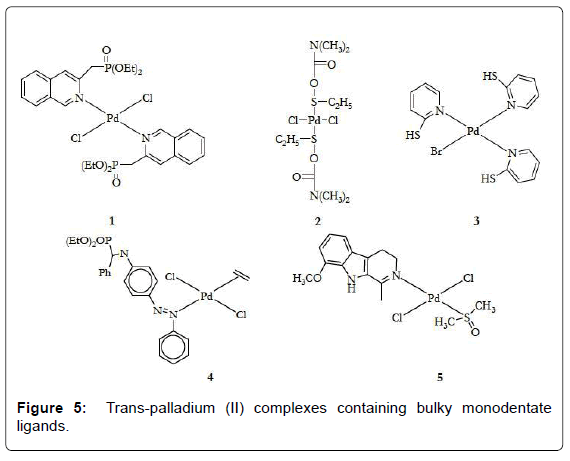
Figure 5: Trans-palladium (II) complexes containing bulky monodentate ligands.
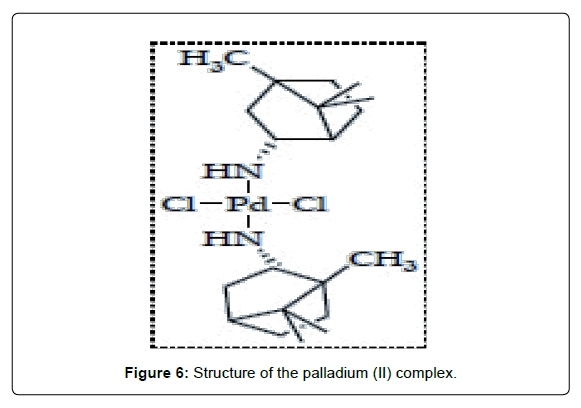
Figure 6: Structure of the palladium (II) complex.
Ruthenium (II & III) Complexes as Antitumor Agents
Recently it was shown that Ruthenium (Ru) possesses several favorable chemical properties, suggesting that it may be a strong candidate to replace platinum. Ruthenium is less toxic than platinum and it is believed that the anticancer activity of ruthenium resides in its ability to mimic iron in binding to several biomolecules, including serum transferrin and albumin. Two Ru(III) complexes, namely trans- [RuCl4(Im)(DMSO)]ImH (NAMI-A) (Figure 7- complex 1) and trans- [RuCl4(Ind)2]IndH (KP1019) (Figure 7- complex 2) are undergoing clinical trials. KP1019 is cytotoxic to cancer cells, NAMI-A is relatively non-toxic but has anti metastatic activity [28-32]. It has been proposed that the activity of Ru (III) complexes, which are usually relatively inert towards ligand substitution, is dependent on in vivo reduction to more labile Ru (II) analogues. Thus, the activity of Ru (II) complexes is currently being explored. In particular, since arenes are known to stabilize ruthenium in its 2+ oxidation state, the potential of Ru (II)–(η6- arene) complexes as anticancer agents is under investigation [31-33]. Half-sandwich (η6-arene)Ru(II) complexes with imidazole, sulfoxide, phosphane, chelating amino acidato, and diamine or diimine ligands have also been evaluated for cytotoxic activity. Both the size of the arene and the lability of the Ru–Cl bond have found to play a crucial role in determining the cytotoxicity of ruthenium (II) complexes of the type [(η6-arene)RuCl(LL´)](PF6). Compounds with extended polycyclic arenes (e.g. tetrahydroanthracene) and LL´ = ethylenediamine (en) are most active towards A2780 human ovarian cancer cells, whereas those with polar substituents on the arene such as COOCH3 (an electronwithdrawing group) or with aromatic diimine ligands such as 2,20-bipyridine (bpy) or 1,10- phenanthroline (phen), exhibit either poor or no activity. Further increases in the size of the polypyridyl ligand (pp) lead, however, to a dramatic reversal of the latter trend and the in vitro cytotoxicities of the complexes [(η6-C6Me6)RuCl(pp)] (CF3SO3) (Figure 8) towards the human cell lines HT-29 (colon cancer) and MCF-7 (breast cancer) are strongly dependent on the surface area of the aromatic system. For instance, the IC50 values decreases from 11.1 over 2.12 to 0.13 μM for MCF-7 cells as the size of the polypyridyl ligand increases in the order dpq < dppz < dppn (dpq = dipyrido- [3,2- f:2´,3´- h]quinoxaline; dppz = dipyrido[3,2-a:2´,3´-c]phenazine; dppn = benzo[i]dipyrido[3,2-a:2´,3´-c]phenazine). These value correlate well with the cellular uptake efficiency, which increases from 1.1 over 146.6 to 906.7 ng (Ru)/mg(protein) within the series. The kinetically inert complexes [(η6-C6Me6) Ru{(NH2)2CS}- (pp)](CF3SO3)2 (pp = dppz, dppn) (Figure 9) are also cytotoxic and this suggests that specific properties of the large polypyridyl ligands (e.g. DNA intercalation and/or cleavage) may be responsible for their biological activity. DNA binding studies indicate that (η6- C6Me6)Ru(II) compounds containing dpq or particularly dppz ligands are good metallointercalators but that the dppn ligand is too large to support stable intercalation between the base pairs of the double helix [34-42]. The in vitro and in vivo assessment of a series of η6-arene ruthenium complexes containing a pta ligand (pta = 1,3,5-triaza-7- phosphaadamantane) were evaluated as anticancer agents [44]. In addition to the benzene, toluene, para-cymene and hexamethylbenzene derivatives, three systems with functionalised arene ligands were tested, (Figure 9, complexes 1, 2 and 3). All pta complexes were found to cause pH-dependent DNA damage, in such a way that DNA was damaged at the typical pH of hypoxic tumor cells, whereas little or no damage was observed at characteristic pH values of healthy cells. This behavior was attributed to the pta ligand, which can be protonated at low pH, and the protonated form was considered to be the active agent. Therefore, the introduction of functionalised pendant arms on the arene ligand, such as in the below complexes (Figure 8), did not show any significant improvement in the cytotoxicity of the compounds (IC50 (TS/A cells) = 66 μM,; 103 μM,; 159 μM,) as compared to the η6- aromatic hydrocarbon systems.
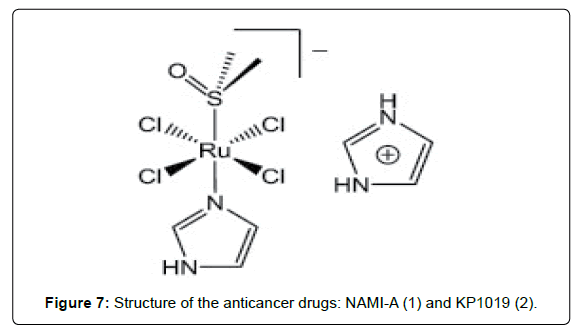
Figure 7: Structure of the anticancer drugs: NAMI-A (1) and KP1019 (2).
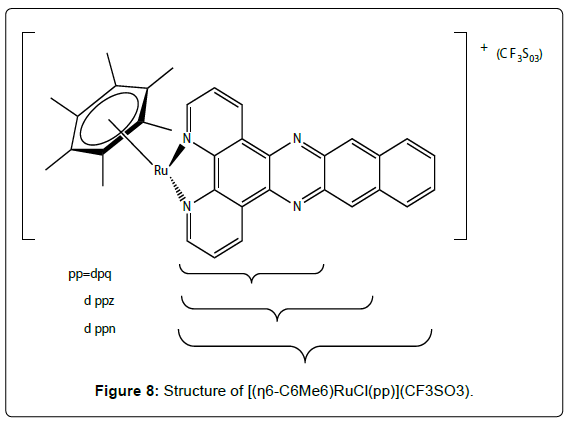
Figure 8: Structure of [(η6-C6Me6)RuCl(pp)](CF3SO3).
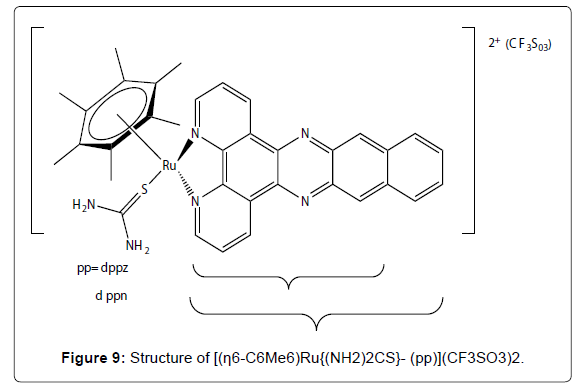
Figure 9: Structure of [(η6-C6Me6)Ru{(NH2)2CS}- (pp)](CF3SO3)2.
Copper Complexes as Antitumor Agents
The anticancer activities of range of simple copper complexes incorporating different types of nitrogen donor ligands such as purine, thiosemicarbazone, imidazole, benzohydroxamic acid and amino acid ligands have been studied [44-47]. Some mixed chelate copper-based drugs have exhibited greater antineoplastic potency than cisplatin in vitro and in vivo studies [48,49]. Copper complexes appear to have a mechanism of action significantly different to that of the clinically used drug cisplatin. For example, the complex [Cu(malonate)(phen)2] was shown to induce apoptosis in cultured mammalian cells and is known to mediate significant cellular oxidative stress, promote membrane lipid peroxidation and interfere with mitochondria respiratory activity in fungal cells [50,51]. Similar copper chelates of phen were studied and also showed high antineoplastic activity by inhibiting respiration and ATP synthesis [52]. Furthermore, [Cu(phen)]2+ type complexes are known to bind to DNA both intercalatively and non-intercalatively and are known as potent oxidative nucleases but the exact structure of [Cu(phen)]2+ when bound to DNA has not been fully characterized [53]. Recently Devereux et al. have been examining alternative chelating ligands such as TBZH and 2-PyBZIMH (Figure 10- complex 1 and 2). The anti-cancer activity of TBZH is enhanced greatly when it is bound to a copper centre [54] but none of the complexes reported so far have exhibited activity close to that of cisplatin. Reports of the biological activity of 2-PyBZIMH are scarce with no papers published describing the anti-cancer activity of its copper complexes. Its use as a ligand in the preparation of novel Pt (II) and Pd(II) anti-cancer agents has been reported [46]. These workers did not test the free 2-PyBZIMH ligand but found that [Pt(2-PyBZIMH)Cl2] was the most efficient anticancer agent against eight brain tumour cell lines, but the activity was significantly less than that of cisplatin. Recently, synthesized a complex of Cu (II) with bis(3 acetylcoumarin) thiocarbohydrazone (Figure 11). This distorted octahedral complex has shown promising cytotoxic activity when screened using the in vitro method for certain types of cell lines. Bravo-Gómez et al. selected a mixed chelate copper (II) complexes of [Cu (N–N)(acac)]NO3 and [Cu(N– N)(gly)]NO3 with several substituents on the diimine ligand to perform a quantitative structure–activity relationship (QSAR) study. Moreover, Chen et al. have synthesized copper (II) complexes of ethyl 2-[bis(2-pyridylmethyl) amino]propionate ligand (ETDPA). These complexes are stable in air with the formula of [(ETDPA)CuCl2] and [(ETDPA)Cu(phen)](ClO4)2. The DNA-binding properties of the two Cu complexes with DNA was different. The binding mode of [(ETDPA)CuCl2] is not the classical intercalation binding, and the binding mode of [(ETDPA)Cu(phen)] (ClO4)2 with DNA is intercalation. The cytotoxic assay shows that the [(ETDPA)Cu(phen)](ClO4)2 is more active on the proliferation of cancer cells than the [(ETDPA)CuCl2].
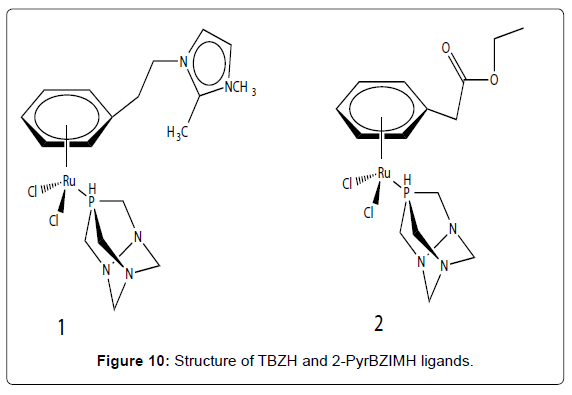
Figure 10: Structure of TBZH and 2-PyrBZIMH ligands.
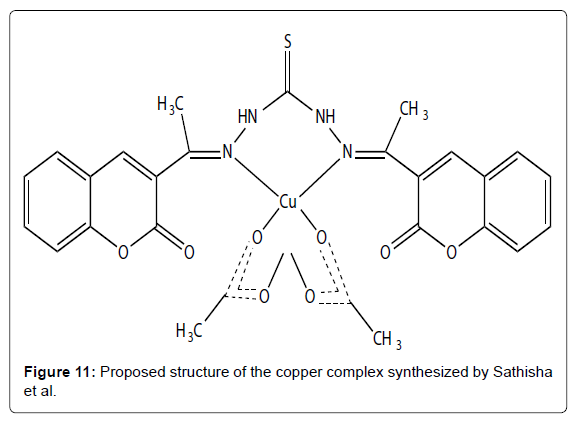
Figure 11: Proposed structure of the copper complex synthesized by Sathisha et al.
Anticancer Agents: Titanium Complexes
Among the metals studied to dates which comprise much of the periodic table complexes of titanium have shown particular promise due to high activity against tumors that are resistant to cisplatin combined with low toxicity [55-62]. In particular, the titanium complexes bis(cyclopentadienide)titanium dichloride [(Cp2TiCl2), titanocene dichloride, TDC, (Figure 12 – 1 complex)] and cis-diethoxybis(1-phenylbutane-1,3-dionato)titanium(IV) [(bzac)2- Ti(OEt)2, budotitane, (Figure 12- 2 complex)] stand out as the first metal-based chemotherapeutics to reach Phase I clinical trials since the development of cisplatin. Although both TDC and Budotitane showed promise in these preliminary studies, TDC has not progressed beyond Phase II due to its low efficacy vs. toxicity ratio, [63,64] and Budotitane has not progressed past Phase I due to formulation problems [65]. These difficulties have boosted the development of titanium complexes that display higher activity and hydrolytic stability. The strong cytotoxic activity of TDC against implanted Ehrlichascites tumors (EAT) in mice was reported, where cure rates greater than 80% were observed with little of the heavy-metal toxicity observed with cisplatin [66]. In subsequent studies the authors reported the key observation that while TDC and vanadocene dichloride displayed similar activities against EAT in vivo and on MCF-7 in vitro , the cytotoxicity of TDC was 100 times less than that for vanadocene dichloride in vitro [67,111]. The reason for this drop in activity was hypothesized to be due to the hydrolytic instability of TDC, which rapidly forms insoluble aggregates in aqueous solution with eventual formation of TiO2, a feature that is known to occur with many Ti (IV) complexes due to their electron-poor and oxophilic nature. A detailed study of the hydrolytic behavior of TDC in neat water at various pH ranges revealed that at pH 5, following a rapid loss of the first chloride ion, complete hydrolysis of both chloro groups occurs with a t1/2 of ca. 50 min, while hydrolysis of the Cp ligands is slower (t1/2 ca. 54 h). In contrast, vanadocene dichloride was stable toward hydrolysis at pH 7, which likely explains the difference between the in vitro and in vivo results for these two compounds. The in vitro cytotoxicity could be restored by employing a galenic formulation, and a subsequent patent reported a water-soluble formulation of TDC, MKT-4, which has allowed for its intravenous administration during clinical trials. The anticancer properties of Budotitane were tested in Phase I Clinical Trial [74]. It possesses activity towards animal tumors such as EAT and colon tumors [56,65]. When examining its hydrolysis, addition of Budotitane to neat water resulted in a suspension where the complex remained completely undercomposed. However, if water was added to a solution of Budotitane in methanol or acetonitrile, rapid hydrolysis ensued. The half-life for hydrolysis of the ethoxy groups in water was calculated as t1/2 = 20 s, while hydrolysis of the diketonato ligand in acetonitrile/water solutions of Budotitane is observed after 2.5 h. The use of smaller diketonato ligands was noted to lead to even more rapid ligand hydrolysis. NMR studies indicate that Budotitane exists as the cis isomer [56]. MM3 studies suggest that the predominance of the cis compound is primarily due to steric factors. In an interesting demonstration of the additional structural complexities that can accompany the use of transition metals as drugs, the unsymmetrical ligands of Budotitane can potentially give rise to three sets of enantiomeric cis complexes. In CDCl3 solution at 23 °C, Budotitane exists as a 4:1 mixture of two cis isomers. The relative cytotoxicity of these two compounds has not been investigated, although the crystal structure of one of them has been determined [69].
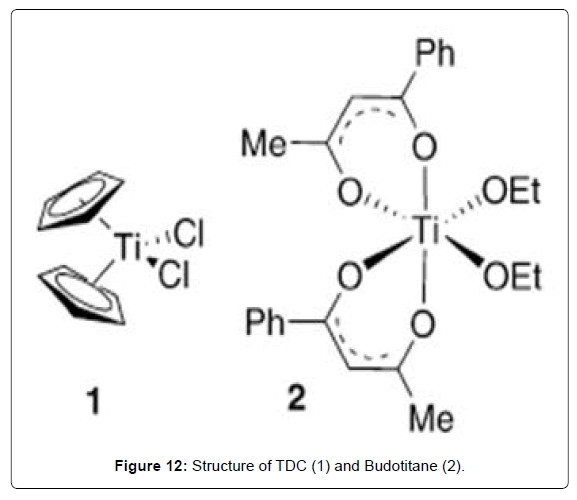
Figure 12: Structure of TDC (1) and Budotitane (2).
Structure–Activity Relationships of Titanocene Dichloride and Related Compounds: Many work has been performed in developing therapeutic analogues of TDC by varying the central metal (M), the labile ligands (Cl) and the bis-cyclopentadienyl moiety.The study of cytotoxic metallocenes other than titanium has been described in detail elsewhere [70]. Substitution of the chloro ligands has been explored in order to produce analogues with greater water solubility, but in general has not been shown to have a strong impact on cytotoxic activity. For example, titanocenes of the general formula Cp2TiX2 (X = Cl, Br, NCS, maleate, O2CCl3) all achieved cure rates of 100% in mice with fluid EAT, consistent with the hypothesis that the cytotoxic Ti (IV) species is generated from displacement of the labile halide ligands in TDC [71]. In contrast, small changes to the Cp ligand can strongly affect the hydrolytic stability and water solubility properties of the metallocenes and thereby impact the cytotoxic activity. In addition, it has been proposed that substitution can also affect the ability of the ligand to intercalate with DNA, although further work needs to be done to establish this mechanism. Substitution of one of the hydrogen atoms of the Cp ligand by R [R = Et (Figure 13 – complex 3), R = Si(CH3)3 (Figure 13- complex 4)] was reported to reduce cure rates of mice with fluid EAT to 60– 80%, while substitution of both Cp ligands by R reduced dramatically to 10–30% the survival [55]. Pentamethyl-substituted titanocenes were reported to be completely devoid of cytotoxic activity. The extent to which this lowered activity represents the reduced water solubility of these compounds is uncertain. Studies on the hydrolytic behavior of the methylated metallocene compound (C5H4Me)2TiCl2 (Figure 13-complex 5; C5H4Me = 1-methylcyclopentadienide) complexes revealed that the CpMe ligands were less readily hydrolyzed than TDC at physiological pH. Furthermore, when the chloride ligands were replaced with glycine (Figure 13 - complex 6), water-soluble complexes with high stability at physiological pH were obtained. The complexes were observed to form stable complexes with nucleotides at pH 7, although further biological studies were not reported [72]. Substitution of the Cp ring by electronpoor substituents (R = CO2Me, Figure 13 complex 1 ) was reported to lead to increased cytotoxicity in a human small-cell lung-cancer cell line but revealed little cytotoxicity towards a number of other cells studied [73]. The latter may be explained by reduced hydrolytic stability due to decreased ligand-to-metal electron donation. A large number of titanocene analogues containing aromatic groups appended to the Cp ligand have also been synthesized [62]. Although generalizations regarding structure– activity relationships are not yet clear, these complexes have shown good in-vitro activity against the LLC-PK cell line. When tested against a panel of 36 human tumor-cell lines, the most promising candidate, titanocene Y (Figure 14 - complex 8), was found to have good activity against renal cell cancer. Interestingly, the oxalato (Figure 14 - complex 9), possessing a substantially less labile chelating ligand in replacement of the chloro groups, was reported to have a 13- fold increase in activity relative to titanocene Y during in vitro studies against the LLC-PK cell line [74]. Yet in vivo studies showed almost identical activity to titanocene Y [75]. Although the hydrolysis behavior of titanocene Y and oxalato was not studied in detail, it is possible that differences in the IC50 values of titanocene Y and oxalato obtained in vitro may in fact reflect the greater hydrolytic stability of oxalato. In general, lower activity was found in analogues where the two Cp units were bound by a two-carbon bridge, obtained as mixtures of stereoisomers (Figure 14- complex 10), although other ansametallocene complexes studied recently revealed promising activity [76,77]. Several attempts have been made to improve the aqueous solubility of TDC through appendage of polar side chains to the Cp ligands. For example, titanocenes with alkylammonium hydrochloride moieties afforded improved aqueous solubility. (Figure 15- 11) showed good activity against both A2780 and the cisplatin-resistant A2780/ CP cell lines [78]. Interestingly, an analogue of the complex 11 (Figure 15- 11) with a bulkier Cp ligand (Figure 15- 12) possessed comparable cytotoxicity [79]. Concurrently, in a parallel study, a significant number of similar alkylammonium complexes were synthesized and their in vitro cytotoxic properties assessed against A549, H209, and A2780 cell lines in direct comparison with the water soluble preparation of TDC, MKT-4 [80]. In initial studies an intriguing difference in cytotoxicity was found between bis(alkylammonium)cyclopentadienide complexes and those containing a single alkylammonium Cp ligand [81]. Subsequent studies on related complexes, however, have found that this “bis(alkylammonium)” effect is not necessarily general. A number of cationic, water-soluble derivatives of TDC have been prepared through incorporation of an ester or a secondary amide on to the Cp ligand [82,83]. By employing an acylation reaction, a diverse array of amide and ester-containing titanocenes were synthesized. In vitro studies of complexes with the general structure of the complex 14 (Figure 16) showed good activity against BJAB (leukemia) cell lines [84]. Notably, complex 15 (Figure 15) possesses a fluorescent aminopyrene unit which may be of potential application in studies on the biological activity mechanism.
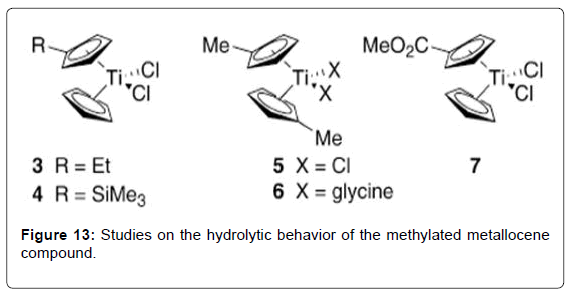
Figure 13: Studies on the hydrolytic behavior of the methylated metallocene compound.
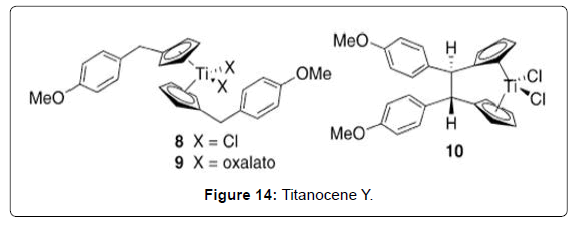
Figure 14: Titanocene Y.
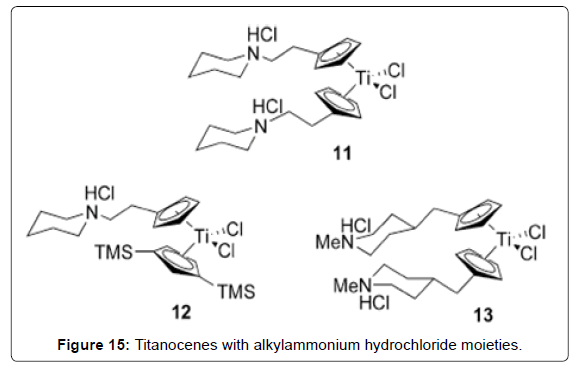
Figure 15: Titanocenes with alkylammonium hydrochloride moieties.
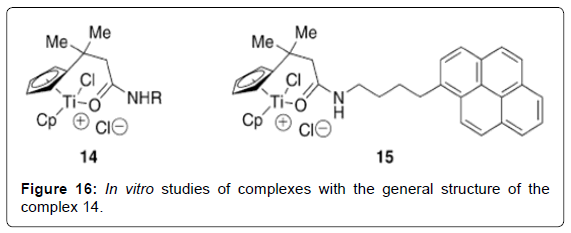
Figure 16: In vitro studies of complexes with the general structure of the complex 14.
Structure–Activity Relationships of Budotitane and Related Compounds: The structure–activity information gleaned from the synthesis of 200 derivatives of budotitane with the general formula D2MX2 (D = 1,3 dicarbonyl;M = metal; X = labile ligand) and their activity against the sarcoma 180 ascitic tumor model were reported. Among the metals investigated, tumor-inhibiting effect decreased in the order Ti>Zr >Hf >Mo >Sn >Ge. No significant difference in cytotoxicity was found between compounds incorporating the labile groups X (X = F, Br, Cl, OEt), although the OEt group was eventually chosen due to its greater hydrolytic stability. Among the 1,3-dicarbonyl groups studied, acetylacetonato (acac) compounds possessed very low activity, while optimal antitumor activity was found for the benzoylacetonato (bzac) ligand. Incorporation of electron-withdrawing (Cl, NO2) or donating (OMe) groups on the aromatic ring had a slightly deleterious effect upon the observed activity, as did the replacement of phenyl for tertbutyl, although considerable cytotoxicity was retained. The cytotoxicity of structurally related mononuclear and polynuclear titanium (IV) 4-acyl-5-pyrazolonato (Figure 17- complex 16) species that are structurally related to Budotitane were explored [57,85]. The cytotoxicity of a tetranuclear species (Figure 17- complex 17) was encapsulated in a liposome due to insufficient water solubility and found to display cytotoxicity against TA-3 and HEP-2 cell lines in vitro. Synthetic studies found that selectivity for mononuclear compounds could be obtained by rigorous avoidance of moisture during their synthesis. Notably, addition of trace water to a solution of the monomeric compound 18 (Figure 17- complex 18) in anhydrous [D6] benzene formed a highly stable species that was hypothesized to be hydrolyzed oxo-bridged oligomers retaining the chelating ligand within 15 min, unlike the observations with budotitane [56].
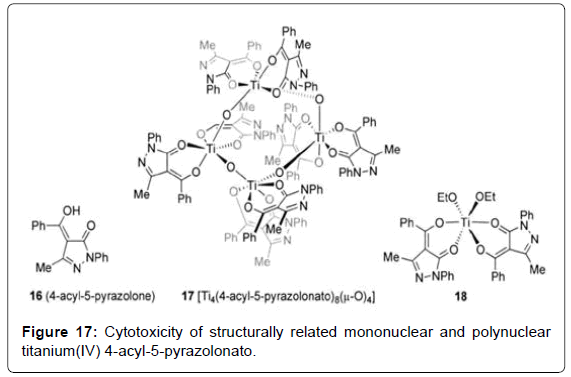
Figure 17: Cytotoxicity of structurally related mononuclear and polynuclear titanium(IV) 4-acyl-5-pyrazolonato.
Comparison of the Biological Profiles of Titanocene Dichloride and Budotitane: In general, the biological activity profiles of TDC and Budotitane show many congruencies. When in vivo studies are compared, TDC and Budotitane show good activity against slowgrowing tumors of the colon, liver, and kidney, and relatively poor activity against faster growing cancers such as leukemia. Notably, emesis and other symptoms associated with the toxicity of cisplatin were absent during clinical trials of TDC and Budotitane. Both compounds hydrolyze easily, and require galenic preparations with water-soluble ligands in order to impart water solubility and stability. Additionally, both complexes contain a set of aromatic ligands that undergo hydrolysis under physiological conditions within several hours. The observation that TDC and Budotitane do not exert their maximal cytotoxic effects until >24 h after administration in stark contrast to cisplatin certainly leads to the question of how this relates to the half life of ligand hydrolysis [85]. In addition, the observation that TDC possess enhanced cytotoxicity following aging in certain organic cosolvents [61] raises questions regarding the particular lability required of active complexes. While a number of detailed biological studies have been performed on TDC, analogous biological studies for Budotitane are largely absent. Although it is reasonable to hypothesize that TDC and Budotitane share a common mechanism of action, too little is known at this stage to draw definite conclusions. The following section will attempt to summarize what is currently known about the mode of action of these cytotoxic titanium complexes.
Mechanistic Aspects of Biological Activity of TDC and Budotitane
Early hypotheses that TDC share a mechanism of action with cisplatin have been challenged by the efficacy of TDC against cisplatinresistant cell lines, as well as by the disparate chemical behavior of “hard”, hydrolytically unstable Cp2TiCl2 with “soft”, water soluble Cl2Pt(NH3)2 observed in aqueous solution under physiological conditions [63]. Nucleic acid metabolism appears to be disturbed by titanocene complexes. Analysis of the intracellular localization of titanium after treatment of TDC found titanium accumulation in cellular regions rich in nucleic acids [88]. Interestingly, while cisplatin immediately causes cessation of DNA-metabolic activity, TDC does not affect cell transit through the S phase but cells are irreversibly unable to perform mitosis [89]. It has been concluded that the three main phenomena observed in cells after treatment with TDC – specifically, the formation of giant cells, the activation of endogenous viruses, and the development of cellular necrosis – are all consistent with an interaction of TDC with intracellular DNA [90]. Adducts of TDC (Cp2Ti–DNA and CpTi–DNA) with calf thymus DNA has been detected spectroscopically,[88] as was also supported by model studies with particular nucleotides [92]. The specific interaction of the active titanium species with DNA remains unclear, although an interaction with the phosphate backbone has been proposed [93]. As far as Budotitane is concerned, the contribution of the aromatic diketonato ligand toward higher cytotoxicity has led to hypotheses involving DNA intercalation [56,85]. However, in light of the considerable activity retained when C6H5 is replaced by tert-butyl, the uncertain nature of the active species, and the paucity of biological studies on Budotitane, this proposal remains speculative. Another work described the interaction of TDC with topoisomerase II, an enzyme catalyzing the opening and rejoining of the DNA phosphate backbone during the cell cycle. TDC was reported to be a potent inhibitor of topoisomerase II, which may also lead to antiproliferative effects [94]. Much recent work has focused on the mechanism by which anticancer titanium complexes are transported into cells. Importantly, the question of whether ligands remain attached to the titanium during transport into the cell or whether the ligands merely serve to protect the titanium center from hydrolysis in the intercellular medium remains an important unanswered question. Uncovering the mode of titanium transport would have important implications for the design of more effective and potent cytotoxic titanium complexes. Noting the increased number of transferrin receptors on the surface of cancer cells and the implication of transferrin in transport of metal ions such as GaIII and RuIII to cancer cells [95,96], it has been proposed that transferrin (Tr) is responsible for uptake of titanium from the intercellular medium [98,99]. Transferrin, the 80 kDa protein responsible for iron transport into cells, is present in the intercellular medium at a concentration of ca. 35 μl and contains two metal binding domains that are about 30% saturated with FeIII in blood plasma. Additional studies have shown that TiIV binds more strongly to human serum transferrin than does FeIII [101,102] UV/Vis and 1H NMR studies support the conclusion that TDC as [Cp2Ti(citrate)2] binds to transferrin with complete hydrolysis of the Cp ligands. After reaction with transferrin for 30 min at pH 7, 1H NMR signals attributed to bound Cp in TDC disappeared, replaced by resonances characteristic of free cyclopentadiene. Interestingly, at low pH <5.5, the titanium was released from Ti2Tr to bind to ATP. In this mechanism, TDC, or its analogues [103,104], could possibly serve as a pro-drug, with the ligands protecting the titanium center from hydrolysis into titanium dioxide in the intercellular medium. Although TDC is completely hydrolyzed by transferrin in the presence of added citrate, the binding of transferrin to the potentially oxo-bridged hydrolysis products of TDC may occur with different rates and/or selectivities. In addition, the water-stable tetranuclear titanium (Figure 18 – complex 20) based on the diketonato analogue maltol ligand (Figure 18- complex 19) does not donate TiIV to transferrin, yet is active against HT-29 cell lines in vitro with values comparable to that of TDC, unlike other titanocene derivatives studied in comparison [105]. Thus, it would appear that further work is necessary in establishing transferrin as the exclusive agent of active transport for cytotoxic titanium complexes. A recent paper proposes an alternate theory for titanium transport to cells through the action of serum albumin (HSA) [106]. HSA, which is present in high concentrations in the intercellular medium (ca. 700 μl), possesses a variety of metal-binding sites and has been shown to bind intact metal complexes such as photosensitizers and contrast agents. NMR studies showed that, in contrast to albuminfree controls, solutions of TDC in the presence of HSA showed no visible signals for either free Cp2TiCl2 or cyclopentadiene after several days. Instead, two proton signals were observed, and proposed to represent TiIV-bound Cp protons. These results strongly suggest that HSA stabilizes the Cp2Ti moiety at physiological pH, and that the extent to which albumin binding stabilizes complexes may be crucial to bioactivity. This was supported by studies on other metallocene analogues [9]. In contrast, binding studies with the dichloro analogue of Budotitane showed less than TDC equiv. of Ti bound to the protein and the presence of free ligand in the NMR spectrum. It was also reported that while transferrin outcompeted HSA for titanium citrate, exchange of Ti from a preformed HSA-Ti complex to transferrin was slow and incomplete. The important distinction of this model is that the ligands of the cytotoxic titanium complex remain bound, allowing for a biological mechanism implicating the ligands in a putative DNAbinding event within the cell. An important unanswered question in this model is whether there is a mechanism by which the HSA-Ti complex is transported into the cell, or whether HSA serves as a protecting group for the active titanium complex until it is transported by passive diffusion into the cell. The discovery that serum albumin can stabilize otherwise hydrolytically unstable titanium complexes has, therefore, added a new dimension to the study of these compounds. It is thus clear that there is significantly more to be studied than is known about TiIV complexes as cytotoxic agents. Despite the many years since the initial findings regarding the involvement of TiIV centers in biological interactions of a therapeutic potential, very little is known about the actual fruitful biological interactions involved in the cytotoxicity mechanism, as well as about the exact requirements of a suitable complex and its attendant ligands. Thus far, only complexes of Cp, diketonato, or related ligands have been explored, although some recent derivatives demonstrate markedly improved features. Nevertheless, the enormous potential of the TiIV center due to its notably low toxicity calls for a reinvestigation of its diverse complexes.
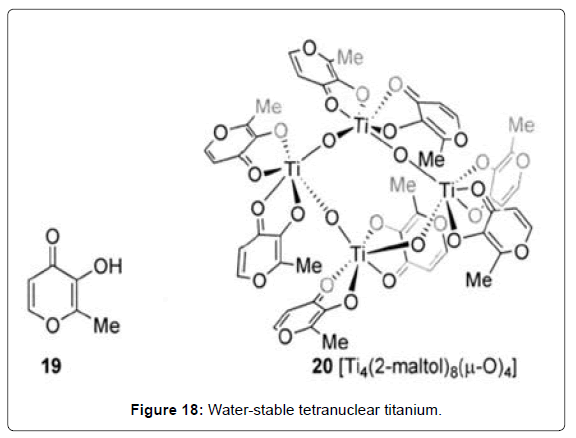
Figure 18: Water-stable tetranuclear titanium.
Phase II trial of titanocene dichloride in advanced renal-cell carcinoma
In Clinical Trial, 14 patients with advanced renal cell cancer (RCC) were recruited into phase II trial. Patients were eligible if they had histologically confirmed RCC, bidimensionally measurable metastatic or locally recurrent unresectable disease, no prior medical antineoplastic treatment, a life expectancy of at least 3 months along with a Karnofsky performance status of above 70%, and adequate renal, hepatic, and bone marrow function. The patients' characteristics are outlined in Table 1. All patients were treated with titanocene dichloride (provided by Medac, Hamburg, Germany) given at 270 mg/m2 on day 1 by i.v. injection. Treatment cycles were repeated every 21 days. Patients who achieved partial or complete responses or stable disease after two cycles were continued on titanocene dichloride until either progression or stabilization of their disease for two cycles. The response to treatment was assessed after two cycles of therapy and every 3 months thereafter according to WHO response criteria [108]. A total of 11 patients received 2 cycles of titanocene dichloride. One patient who presented with stable disease continued the treatment for a further two cycles and one patient died due to tumor progression within 4 weeks. There was no complete or partial response. Three patients (21%) had stable disease (SD) for 1, 3, and 3 months, respectively. Two patients had metastases in the lung; one of them had been nephrectomized. The other patient had bone metastases only. All patients with SD had a good performance status (Karnofsky index 100%). The remaining ten patients (79%) had shown evidence of progression while under treatment with titanocenedichloride. The median survival time of all patients amounted to 50 weeks, and the overall survival was 43% at 1 year. In eight patients (57%) symptoms of nausea, vomiting, and weight loss occurred; three of them suffered from grade 3 WHO toxicity and two patients, from grade 2 toxicity. Subsequently, the dose of titanocene dichloride was reduced to 75% of the original dose in two patients. There was no other organ toxicity (Table 2). Four patients (29%) developed an increase in serum creatinine concentration corresponding to WHO grade 2 toxicity (maximal 2.6 mg/dl) and three patients (21%) developed grade 1 toxicity. In regard to hematologic parameters and chemistry values of liver function, nosignificant difference was detectable between baseline levels and control values during the treatment. In conclusion TDC produced responses in proportions too small to justify its use in singleagent therapy.
| Charecteristics | Number of Patients |
|---|---|
| Eligible Patients | 14 |
| Sex | |
| Male | 11 (79%) |
| Female | 3 (21%) |
| Age(years) | |
| Median (range) | 60 (33-72) |
| Karnofsky index (%) | |
| 80 | 1 (7%) |
| 90 | 9(64%) |
| 100 | 4(29%) |
| Nephrectomy | 10(71%) |
| Sites of metastatic disease | |
| Lung | 3(21%) |
| Lung and others | 4 (29%) |
| Bone | 4 (29%) |
| Lymph nodes | 1 (7%) |
| Local recurrence | 2 (14%) |
Table 1: Patients’ characteristics.
| Side Effects | WHO grade | Total(%) | ||
|---|---|---|---|---|
| I | II | III | ||
| Loss of appetite | 14 | 0 | 0 | 14 (100%) |
| Nausea/vomiting and weight loss | 3 | 2 | 3 | 8 (57%) |
| Increase in Creatinine | 3 | 4 | 0 | 7 (50%) |
Table 2: WHO toxicity of treatment with titanocene dichloride (n=14).
Methoxy alkyl substituted titanocenes
Recently, some of us have reported the synthesis and the cytotoxic activity of some titanocene derivatives, obtained replacing the arylmethoxylic group on cyclopentadienyl of titanocene Y with the ethenylmethoxy group, in order to have a stronger electron donor effect on the cationic species responsible for the cytotoxic activity (Figure 19). We also verified the influence of leaving ligands on the activity by substituting chlorine atoms with dimethylamide, oxalate or aminoacid groups [107]. It is worth noting that in [108], the highest cytotoxic activity was reported for half-titanocene complex T1, compared to titanocene complex T2, titanocene Y and cis-platinum. Some of the synthesized compounds showed a good cytotoxicity, in particular the complexes bis-cyclopentadienyl-ethenylmethoxyl-titanium dichloride T2 and cyclopentadienyl-ethenylmethoxyl-titanium trichloride T1 gave values very similar to cis-platin on MCF-7 cell lines, with the IC50 comparable to the ones reported for titanocene Y. Moreover, the half-titanocene complex T1 also showed a good cytotoxic activity, comparable to that of cis-platin, on HEK-293 cells. The results of the hydrolysis of our titanocenes showed unequivocally that the leaving groups (Cl, N(CH3)2, C2O4 or glycine) significantly affect even the hydrolysis rate of cyclopentadienyl groups, being chloride and oxalate more stable [108]. Thus, the presence of substituents, aryl methoxy group on cyclopentadienyl ring in titanocene Y or ethenyl-methoxy group in titanocene complex T2 or in the half-titanocene complex T1, produces compounds having interesting cytotoxic activity. Although generalizations regarding structure-activity relationships are not yet clear, we could hypothesize that the neutral nucleophilic substituents of cyclopentadienyl (aryl methoxy or ethenyl-methoxy group) could intramolecularly coordinate to the titanium cation, thus preventing decomposition reactions. On the other hand, this hypothesis was suggested for analogous complexes able to give polymerization of propene or styrene having microstructures strongly influenced by the possible coordination of neutral substituent of cyclopentadienyl at the metal center. As mentioned above, in the literature several examples of titanocene-complexes showing cytotoxic activity were reported, but, to the best of our knowledge, the cyclopentadienyl-ethenylmethoxyltitanium trichloride represents the first example of half-titanocene complex with interesting cytotoxic activity under reduced pression.
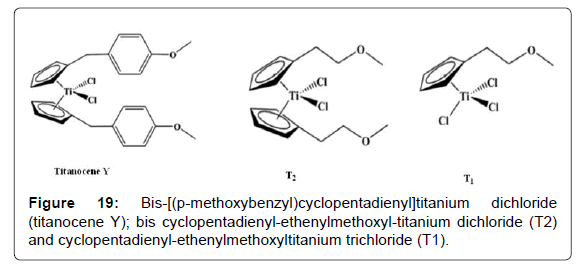
Figure 19: Bis-[(p-methoxybenzyl)cyclopentadienyl]titanium dichloride (titanocene Y); bis cyclopentadienyl-ethenylmethoxyl-titanium dichloride (T2) and cyclopentadienyl-ethenylmethoxyltitanium trichloride (T1).
The earliest reports on the therapeutic use of metals or metalcontaining compounds in cancer show an effective use in clinical practice. They were forgotten until the 1960s, when the anti-tumour activity of the inorganic complex cis-diammine-dichloroplatinum(II) (cisplatin) was discovered. This led to the development of other types of non-organic cytostatic drugs. Cisplatin has developed into one of the most frequently used and most effective cytostatic drugs for the treatment of solid carcinomas. Unresolved problems in platinumbased anti-cancer therapy have stimulated increased research efforts in the search for novel non platinum-containing metal species as cytostatic agents. Preclinical and clinical investigations showed that the development of new metal agents with modes of action different from cisplatin is possible. Thus, complexes with iron, cobalt, or gold central atoms have shown promising results in preclinical studies and compounds with titanium, ruthenium, or Copper central atoms have already been evaluated in phase I and phase II trials. Also, we want propose the organo-metallic complex and modification as new candidates in chemotherapy.
The authors are grateful to the University of Salerno for funding with funds from the University FARB.
The authors would like to thank Massimiliano Spinelli Data Manager of S.S.D. Sperimentazione animale, from ISTITUTO NAZIONALE PER LO STUDIO E LA CURA DEI TUMORI “FONDAZIONE GIOVANNI PASCALE” – IRCCS - ITALIA, for kind help in providing informatic assistance.