Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Research Article - (2014) Volume 3, Issue 1
An efficient and convenient method was developed for the one-step synthesis of various substituted ethyl 2-(1H-indol-2-yl) acetates via a palladium-catalyzed regioselective cascade C-H activation reaction. Importantly, this practical approach can be carried out with readily accessible starting materials and exhibits excellent functional group compatibility.
Keywords: Palladium
2-substituted indoles are important structural motifs present in diverse biologically active molecules [1] and are precursors for a wide variety of alkaloids, such as vindoline [2], vindorosine [2], ellipticine [3], etc. Among them, 2-(1H-indol-2-yl)acetic acids constitute a valuable class of building blocks for natural product and natural product analogue syntheses (Figure 1) [4-7], combinatorial [8], diversity-oriented syntheses [9,10], and medicinal chemistry [11-18]. In addition, they can serve as highly attractive precursors for various chemical transformations, such as diazomethylation to 1-diazo-3-(2- indolyl)-2-propanone [19] and reduction to 2-(2-hydroxyethyl) indole [20]. Therefore, the development of efficient synthetic methods for these compounds has received much attention.

Figure 1: Examples of 2-(1H-indol-2-yl) acetic acids-related natural products and natural product analogues.
Hydrolysis of ethyl 2-(1H-indol-2-yl) acetates is an important route to 2-(1H-indol-2-yl) acetic acids. Accordingly, the literature describes several preparations of ethyl 2-(1H-indol-2-yl) acetates. For example, Capuano et al. demonstrated that the intramolecular Wittig reaction of 2-[(ω-alkoxycarbonylacyl) amino] benzyltri-phenylphosphonium salts produced 2-(1H-indol-2-yl)-acetate with a 78% yield [21]. Furthermore, Moody et al. reported that reductive cyclisation of the ethyl 4-(2-nitropheny)-acetoacetate using titanium(III) chloride in aqueous acetone gave 2-(1H-indol-2-yl) acetate with a 75% yield [22]. Despite producing good yields, these methods suffer from indispensable multi-step pre-transformations of commercially available starting materials, and are therefore of limited synthetic scope [23]. Wilkens et al. described a one-pot reaction of N-phenylhydroxylamine, benzaldehyde and ethyl 2,3-butadienoate followed by hydrolysismediated production of 2-(1H-indol-2-yl)acetate [24]. In spite of a 49% yield, this approach also had its drawbacks, including the use of unstable and relatively rare reactant (i.e., allenes) that might limit their broad applications. Osornio et al. and Guerrero et al. demonstrated that the direct intermolecular oxidative radical alkylation of indole under xanthate-mediated radical conditions afforded 2-(1H-indol-2- yl) acetate in 60% yield [25,26]. Nevertheless, the radical mechanism of this approach might limit its broad synthetic applications.
If indoles can be directly functionalized at the C2-position, the preparation of the target compound class would be more facile. Owing to the development of the transition-metal catalyzed C-H activation chemistry, methods for regioselective direct C2/C3-alkenylation [27-33], alkynylation [34-41], cyano [42,43] and arylation [44-52] of indole nucleus have been well developed to date. However, regioselective C2- alkylation of indole is more challenging.
The Catellani reaction, a palladium-catalyzed norbornenemediated cascade reaction, has been modified to achieve the direct functionalization of indoles [53-55]. Recently, Bressy et al. reported an intramolecular direct arylation of indoles based on modified Catellani conditions [56]. More recently, Jiao et al. reported a direct 2-alkylation reaction of indoles relying on a Pd(II)-catalyzed norbornene-mediated direct alkylation method for indoles, which regioselectively installs an alkyl group to the C2-position of free N-H indoles [57,58]. However, the original publication did not report the use of 2-bromoacetate as an alkylating reagent. To the best of our knowledge, a straight-forward approach for direct C2- alkoxycarbonylalkylation of indoles has not yet been well established. Given its high C2-regioselectivity and excellent functional group tolerance, we envisioned to apply Bach’s method to the synthesis of 2-(1H-indol-2-yl) acetates by employing indoles and ethyl 2-bromoacetate as the starting materials. As part of our continuing effort to assemble indole-based drug scaffolds [59-61], we here present our findings on one-step synthesis of various substituted ethyl 2-(1H-indol-2-yl)acetates via a palladium-catalyzed regioselective cascade C-H activation reaction. Various substituents are tolerated in this system in moderate to good yields. Our protocol highlights a facile one-step transformation from easily available starting material and excellent functional group compatibility.
Unless otherwise noted, the reagents (chemicals) were purchased from commercial sources, and used without further purification. Water was deionized before used. Analytical thin layer chromatography (TLC) was HSGF 254 (0.15-0.2 mm thickness). Compound spots were visualized by UV light (254 nm). Column chromatography was performed on silica gel FCP 200-300. NMR spectra were run on 300 or 400 MHz instrument. Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (LRMS and HRMS) were measured on spectrometer.
General procedure for synthesis of Ethyl 2-(1H-indol-2-yl) acetates
A vial equipped with a magnetic stir bar and a rubber stopper was charged with Pd(PhCN)2Cl2 (65 mg, 0.171 mmol), indole substrate (1.71 mmol), norbornene (321 mg, 3.41 mmol), NaHCO3 (574 mg, 6.84 mmol) and capped with septa. The vial was evacuated and backfilled with argon and the process was repeated three times. A solution of water in DMF (0.5 M) was added via syringe as the solvent. Under argon, ethyl bromoacetate (0.41 mL, 3.41 mmol) was added via syringe, and then the resulting mixture was stirred at room temperature for 10 minutes. After that, the reaction mixture was then placed in a preheated oil bath at 70°C for appropriate time and vigorous stirring was applied. The reaction was monitored by TLC. Upon completion, the reaction mixture was cooled to room temperature, diluted with ethyl acetate, washed with water (twice) and brine (once), dried over Na2SO4, and concentrated. The residue was directly submitted to flash column chromatography (by dry loading) to afford the ethyl 2-(1H-indol-2-yl) acetates products as yellow oils.
We initiated our study by reacting indole with ethyl bromoacetate in the presence of norbornene and Pd(MeCN)2Cl2. However, the direct application of the reported optimized reaction conditions yielded diethyl 2,2’-(1H-indole-2,3-diyl) diacetate, a 2,3-disubstitued-indole product, as the major product.
According to the postulated catalytic cycle of Bach’s approach [57,58], and distincts from the previous Catellani reaction, the arylpalladium(II) species in the terminating step undergoes protodepalladation (i.e. hydrogenolysis), instead of a further palladium catalyzed carbon functionalization, such as Heck, Suzuki and Sonagahira reactions. Consequently, we speculated that for more active alkyl halide substrates, such as ethyl bromoacetate, it might be possible to quench the further substitution reaction and improve the yields of mono-2-substitued-indole products by the use of different combination of solvent and base. Furthermore, we postulated that the phosphine ligands might have an effect on the rate of the substitution process as well.
With the aforementioned considerations in mind, we screened different reaction conditions for optimal results (Table 1). We conducted the first runs (Table 1, entries 1-6) employing the reported optimized Pd source [Pd(OAc)2], base (K2CO3), reaction temperature (70°C) and time (14 hours) in different solvents (Table 1, entries 1-3). Notably, the higher water content in the solvents was found to significantly increase the selectivity for the mono-2-substituedindole product 1 (Table 1), but led to a compromised conversion rate. These results suggest that water might serve as the hydrogen source in the catalytic system, which accelerates the norbornene-mediated cascade reaction by hydrolysing the final arylpalladium(II) species. As DMF/5 M water produced a better yield than the other solvents, it was selected as the solvent in the following tests. The effect of the Pd source was then investigated (Table 1, entries 7-10). Palladium species other than Pd(MeCN)2Cl2 and Pd(PhCN)2Cl2 were substantially less effective. Further screening of catalysts did not lead to better yields and demonstrated that the reaction may not proceed well in the presence of phosphine ligands which may reduce the palladium(II) species to palladium(0) species and interferes the normal palladium(II-IV) catalytic cycle [62]. Pd(PhCN)2Cl2 was chosen as the preferred catalyst because it provided higher conversion rates than the other catalysts. Given the possibility that excessive water in the solvent might hydrolyse the arylpalladium(II) species formed in the first oxidative addition step and lower the solubility of the intermediates, resulting in a cessation of the catalytic cycle and undesired conversion rate, we switched the solvent back to DMF with 0.5M H2O. Finally, a brief screen of bases showed an interesting correlation between the alkalinity of the base and the yield of the 2,3-disubstitued-indole byproduct 2 (Table 1, entries 11-15). Specifically, the yields of the 2,3-disubstitued-indole products increased significantly as the alkalinity of the base increased, with NaHCO3 affording the target products in desirable yields. On the other hand, insufficient basicity led to undesired conversion rates (Table 1, entries 14 and 15). Thus, DMF with 0.5M H2O as the solvent, NaHCO3 as the base, a reaction temperature of 70°C, and a reaction time of 14 hours were selected as the optimal conditions. It was noteworthy that in all tests, the other possible substituted byproducts, N- and 3-substituted products 3, 4 (Table 1) were not detected.
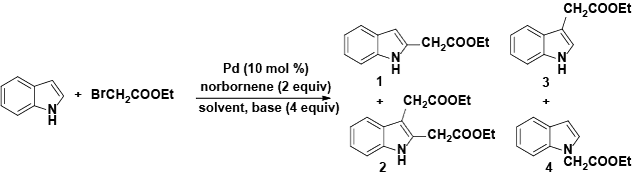 |
|||||
| Entry | Pd Source | Solvent | Base | ||
|---|---|---|---|---|---|
| Yield (%)b | |||||
| 1 | 2 | ||||
| 1 | Pd(OAc)2 | DMA / 0.5 M H2O | K2CO3 | tracec | 56 |
| 2 | Pd(OAc)2 | DMF / 0.5 M H2O | K2CO3 | 7 | 62 |
| 3 | Pd(OAc)2 | DMA / 5 M H2O | K2CO3 | 19 | 26 |
| 4 | Pd(OAc)2 | DMF / 5 M H2O | K2CO3 | 34 | 0 |
| 5 | Pd(OAc)2 | DMF / 2 M H2O | K2CO3 | 24 | 10 |
| 6 | Pd(OAc)2 | CH3CN / 5 M H2O | K2CO3 | 15 | 21 |
| 7 | Pd(Ph3P)2Cl2 | DMF / 5 M H2O | K2CO3 | tracec | 0 |
| 8 | Pd(dppf)2Cl2·CH2Cl2 | DMF / 5 M H2O | K2CO3 | tracec | 0 |
| 9 | Pd(MeCN)2Cl2 | DMF / 5 M H2O | K2CO3 | 46 | 0 |
| 10 | Pd(PhCN)2Cl2 | DMF / 5 M H2O | K2CO3 | 52 | 0 |
| 11 | Pd(PhCN)2Cl2 | DMF / 0.5 M H2O | K2CO3 | 8 | 79 |
| 12 | Pd(PhCN)2Cl2 | DMF / 0.5 M H2O | KHCO3 | tracec | 67 |
| 13 | Pd(PhCN)2Cl2 | DMF / 0.5 M H2O | NaHCO3 | 85 | 7 |
| 14 | Pd(PhCN)2Cl2 | DMF / 0.5 M H2O | KOAc | 58 | 0 |
| 15 | Pd(PhCN)2Cl2 | DMF / 0.5 M H2O | NaOAc | 42 | 0 |
aGeneral reaction conditions: Pd (0.171 mmol), indole (1.71 mmol), norbornene (3.41 mmol), base (6.84 mmol), ethyl bromoacetate (3.41 mmol), solvent (8 mL, total), 70°C, 14 h. bIsolated yield. cMonitored by LC-MS and TLC
Table 1: Optimization of the catalysis conditionsa.
Encouraged by these initial results, we next proceeded to examine the general utility of the Pd(PhCN)2Cl2/norbornene catalytic system for synthesis of a wide range of ethyl 2-(1H-indol-2-yl) acetates (Table 2). Indoles with electron-donating and electrondrawing substituents at the various positions smoothly participated in the C2-ethoxycarbonylmethylation reaction. Four regioisomeric methylindoles effectively participated in the cascade reactions to afford equally good levels of yields (Table 2, entries 1-4). The reaction worked well with different halogens on various positions on the benzene ring (Table 2, entries 6-12), which has three main advantages. First, halogen substituents could be used to adjust the polarity, lipophilicity, and metabolic stability of dyes or pharmaceuticals. Second, halogens allow further modification using cross-coupling reactions for the elaboration of molecule libraries. Third, halogen substituents on various positions can serve as invaluable building blocks for enrichment of Structure- Activity Relationships in medicinal chemistry. It was noteworthy to mention that 5-iodoindole, a potential substrate for classic Catellani reaction, was readily tolerated in our catalytic system and gave the desired product 6l in good yield. Interestingly, electron-deficient indoles reacted better than their electron-rich counterparts to give good yields of 2-(1H-indol-2-yl) acetates under the optimal conditions (Table 2, entries 6-15). Moreover, the reaction could be extended to functionalized azaindoles to give the expected product 6o in modest yield. We also noted that a more bulky 3-position blocked the substrate, 3-methyl-indole, produced the desired product 3 in a 35% yield (Table 2, entry 16). Control experiment showed that this catalytic system did not work in the absence of norbornene. By contrast, N–methylindole failed to participate in the reaction and remained mostly unchanged even after extension of reaction time (Table 2, entry 17). These results are consistent with Bach and Jiao’s latest work regarding the mechanism of regioselective Pd-catalyzed, norbornene-mediated alkylation of indoles. It has been proved that N1-norbornene type palladacycle rather than originally proposed C3-norbornene type palladacycle is formed as the key intermediate in this catalytic cycle [58].
 |
|||
| Entry | R1 | Product | Yieldb |
|---|---|---|---|
| 1 | 4-Me (5a) | 6a | 67% |
| 2 | 5-Me (5b) | 6b | 71% |
| 3 | 6-Me (5c) | 6c | 69% |
| 4 | 7-Me (5d) | 6d | 65% |
| 5 | 5-OMe (5e) | 6e | 80% |
| 6 | 4-F (5f) | 6f | 75% |
| 7 | 5-F (5g) | 6g | 83% |
| 8 | 6-F (5h) | 6h | 82% |
| 9 | 7-F (5i) | 6i | 78% |
| 10 | 5-Cl (5j) | 6j | 85% |
| 11 | 5-Br (5k) | 6k | 84% |
| 12 | 5-I (5l) | 6l | 71% |
| 13 | 5-CN (5m) | 6m | 89% |
| 14 | 5-NO2 (5n) | 6n | 91% |
| 15 | 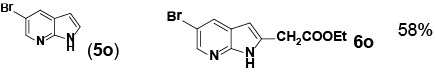 |
||
| 16 | 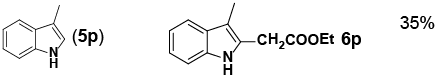 |
||
| 17 | 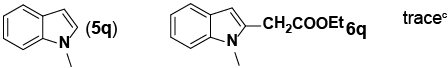 |
||
Table 2: Palladium-catalyzed synthesis of various ethyl 2-(1H-indol-2-yl) acetatesa.
On the basis of above observations, recent publications on the mechanism of the Catellani reaction [63] and extensive mechanistic work by Jiao et al. [58], a plausible mechanism for the observed reaction is proposed in Scheme 1. The norbornene-mediated cascade C-H activation process proceeds as follows: a. N1-position direct palladation; b. syn-aminopalladation of norbornene; c. irreversible palladacycle formation, leading to C2-position ortho-C−H palladation; d. oxidative addition with an alkyl halide to generate palladium IV species; e. reductive elimination of the palladium IV species; f. norbornene expulsion; and g. release of the 2-alkyl indole product and regeneration the Pd(II) species (Scheme 1). The 2-monosubstituted product of this catalytic cycle might undergo further electrophilic substitution process [56] to afford the 2,3-disubstitued-indole product.

Scheme 1: Postulated Catalytic Cycle for a Direct 2-alkoxycarbonylalkylation of Indole by a Norbornene-Mediated Cascade C-H Activation.
To conclude, a practical Pd(PhCN)2Cl2/norbornene catalytic system for the versatile and one-step synthesis of ethyl 2-(1H-indol- 2-yl)acetates has been developed. This practical method features the ability to rapidly and efficiently synthesize various substituted ethyl 2-(1H-indol-2-yl)acetates as precursors to a class of valuable synthetic building blocks, 2-(1H-indol-2-yl)acetic acids. In addition, the protocol exhibits excellent functional group compatibility, leading to valuable derivatives that are not readily available by conventional methods. The present reaction also enables a new access to 2,3-dialkoxycarbonylalkylated indole derivatives which can be applied to indole fused heterocycles and indole alkaloid synthesis.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grants 21021063, 91229204, and 81025017), National S&T Major Projects (2012ZX09103101-072, 2012ZX09301001-005, and 2013ZX09507-001), Sponsored by Program of Shanghai Subject Chief Scientist (Grant 12XD1407100).