Journal of Antivirals & Antiretrovirals
Open Access
ISSN: 1948-5964
ISSN: 1948-5964
Review Article - (2013) Volume 5, Issue 1
Sustained HIV suppression depends on a number of factors including therapy adherence, management of side effects, viral resistance and individual characteristics of patients and therapeutic settings. Treatment response rates range up to 90% in therapy naïve patients but decline to approximately 50% in patients who received several antiretrovirals during treatment history. Furthermore, HIV protease inhibitors (PI) and non nucleoside reverse transcriptase inhibitors (NNRTI) plasma concentrations display high inter- and intra individual variability and the therapeutic window is comparably narrow. In this therapeutic setting the personalization of dosing regimens has been suggested in many cases to tailor the ARV plasma concentrations with the intention to maximize therapy success and minimize side effects in the individual. However, personalizing therapy by modifying the dosing regimen bears the danger of losing therapeutic efficacy, increasing side effects or causing viral resistance.
This topical review identifies pharmacokinetic and pharmacodynamic models of antiretroviral therapy appraising the potential application to HIV therapy and discusses its future in the light of new drug classes and fix-dose combinations.
Keywords: Pharmacodynamic; HIV therapy; Antiretrovirals
The introduction of highly active antiretroviral therapies (HAART) including protease inhibitors and non-nucleoside reverse transcriptase inhibitors dramatically decreased the mortality of AIDS since 1996 [1,2] but also made apparent possibilities and limitations of antiretroviral therapy. Sustained viral suppression depends on a number of factors, which have to be controlled by the patient and physician. (i) HIV therapy only is effective over a longer period of time, if taken continuously and adherently by the patient. (ii) The management of numerous side effects, such as haematological abnormalities, dyslipidemia, polyneuropathy, mitochondrial toxicities, insulin resistance, organ toxicities and lipodystrophy, to mention only a few, is a challenge for physicians. (iii) Viral resistance and cross resistance within all classes of antiretroviral drugs are complicating the situation and lead to therapy failures increasing with the duration of HAART.
Treatment response rates range from 70-90% in therapy naïve patients but decline to approximately 50% in patients who received several HAART during treatment history [3].
HIV protease inhibitors and non nucleoside reverse transcriptase inhibitors (NNRTI) plasma concentrations display a high inter- [4-8] and intraindividual variability [9-11]. The therapeutic window is comparably narrow as drug concentrations expected to be toxic are only 3-5 fold higher than antivirally effective concentrations in vivo. This is a therapeutic setting that suggests establishing personalized dosing regimens to tailor the ARV plasma concentrations with the intention to maximize therapy success and minimize side effects in the individual. In fact, therapeutic drug monitoring of antiretroviral drugs has been suggested by various authors and guidelines [12-23].
However, data and advice for clinicians on the individualization of HAART is rather rare. And personalizing therapy by modifying the dosing regimen bears the danger of losing therapeutic efficacy or increasing side effects. It is good clinical pharmacological practice to base personalized dosing on quantitative information about the relationship between patient’s individual variables, viral resistance patterns, co-administered drugs and plasma concentrations, and between plasma concentrations and therapeutic effects.
This topical review identifies pharmacokinetic and pharmacodynamic models of antiretroviral therapy appraising the potential application to HIV therapy.
Target plasma concentrations in HAART naive patients
Once an effect versus plasma concentration relationship has been established, methods such as therapeutic drug monitoring up to population pharmacokinetics [24] are available to individually adapt the dosing regimen. Target plasma concentrations as minimum effective concentrations (MEC) of protease inhibitors and NNRTI have been defined and extrapolated from studies in therapy-naïve patients [25,26] or in vitro data. Three interventional studies used a similar minimum indinavir concentration threshold, ranging from 0.10-0.15 μg/mL (ATHENA and GENOPHAR studies) [27-29]. The GENOPHAR study also defined in vivo Cmin-thresholds for ritonavir (>2.1 μg/mL), amprenavir (>1.0 μg/mL), lopinavir (>3.0 μg/ mL) and saquinavir (250 ng/mL) [29]. Four studies set the nelfinavir Cmin threshold between 0.52 and 1.0 μg/mL. All studies based their threshold recommendations on the protein-binding adjusted IC50 or IC95-values. Two studies defined an efavirenz Cmin-threshold of >1.0 μg/ mL and an optimum AUC of >60.0 μg×h/mL [30,31] on basis of the data, obtained from the registrational studies with efavirenz [32]. One study found a better virologic response to nevirapine therapy at plasma Cmin concentrations of >4.3 μg/mL [33].
A considerable number of observational studies showed a correlation between drug plasma concentrations of PIs and virological suppression prospectively followed up in phase II studies (saquinavir, indinavir, amprenavir, darunavir). The same has been shown for treatment-naive patients, either in phase III studies (saquinavir, nelfinavir, ritonavir) or commencing HAART in clinics (saquinavir, nelfinavir, indinavir, ritonavir) [34,35].
In general the association between drug concentrations and virological response varies and is less clear in therapy experienced patients.
Target plasma concentrations in treatment experienced patients
Several studies addressed the question whether a correlation of virological and clinical data to pharmacokinetics predicts therapy response on protease inhibitors in extensively pretreated patients. In order to sufficiently and sustained suppress a resistant virus, it can be necessary to achieve high plasma concentrations of antiretrovirals, governed by the viral phenotype. As phenotypic testing is not part of diagnostic routine, these studies emphasized on the combination of virological genotype with pharmacokinetic parameters. Thus, the genotypic inhibitory quotient (GIQ) is the ratio of the trough concentration of antiretroviral agents to the number of resistance mutations detected in the viral genotype. This concept has been confirmed for lopinavir, atazanavir, amprenavir and saquinavir and other protease inhibitors in a number of clinical trials with therapyexperienced patients [36-42].
A. Common two step approaches were used in most evaluations of HAART pharmacokinetics. Data assessed in clinical settings were either analysed in non-compartmental models or fitted to onecompartment analyses. Thus, mean drug concentrations to be expected in diverse populations were evaluated.
B. Population pharmacokinetic approaches tried to evaluate and quantify the factors of influence on HAART pharmacokinetics applied to different populations [43]. Population pharmacokinetics has been used to explore and define relevant cofactors for variation in drug exposure and response in patient populations. Up to date, population pharmacokinetic analyses of more than twenty available antiretrovirals have been published [44-60].
C. Pharmacokinetic/pharmacodynamic models have been used to characterize the (i) relationship between drug exposure and virological and immunological response, and to predict clinical outcome. Modelling and simulation approaches have evaluated (ii) antiretroviral agent outcomes incorporating problematic design and analysis factors, i.e. sparse plasma sampling, data imbalance and censored data. Additional population modelling approaches include (iii) the assessment of dosing compliance, understanding and quantifying drug-drug interactions in order to select dosing regimens and the screening of new drug candidates.
Although these models offer an opportunity for individualizing and optimizing patient therapy, particularly when adjusted for adherence/ compliance, the impact of population pharmacokinetics on clinical antiretroviral therapy is rather restricted, except its contribution to the current regulatory environment, specifically in the area of accelerated approval of new antiretroviral agents.
Population pharmacokinetic studies
Population pharmacokinetic models for nelfinavir detected a number of variables influencing significantly drug concentrations in patients. Very young age, pregnancy and comedication were the three main reasons for a very high variability of values. Neither body weight, age, sex, race, dose level, baseline viral load, metabolite-to-parent drug plasma concentration ratio, history of liver disease, nor elevated results of liver function tests appeared to be significant covariates for nelfinavir clearance [48-50].
The individual indinavir clearance in patients was only decreased by the concomitant intake of the pharmacoenhancer ritonavir, but not other demographic or clinical covariates [47]. The same counts for amprenavir, atazanavir and lopinavir [44,45,56]. Efavirenz and nevirapine plasma concentrations of the two currently used NNRTI in HIV therapy were found to be correlated with impaired liver function and ethnicity [54,55,61]. Higher NNRTI levels in women or Asian patients and a higher efavirenz clearance in white Americans compared to African Americans or non-white Hispanics were detected: cytochrome genetic subtypes influence the pharmacokinetics of NNRTI significantly and were also found to be correlated with therapy outcome [62].
As physiological changes associated with pregnancy have a large impact on the pharmacokinetics of many drugs, a nelfinavir population study analysed the large inter-subject variability 133 HIV- 1 infected pregnant and nonpregnant women [48]. The population pharmacokinetic model described the concentration time course of nelfinavir and its metabolite M8, whereas individual characteristics, such as age, body weight, and weeks of gestation or delivery, were investigated. During late pregnancy, significant increases in nelfinavir (44.4 liters/h) and M8 (5 h(-1)) elimination but unchanged nelfinavir transformation clearance to M8 were observed and nelfinavir clearance showed a twofold increase on the day of delivery, suggesting a decrease in bioavailability on this day. The Bayesian individual pharmacokinetic estimates suggested that the dosage should not be changed in pregnant women but may be doubled on the day of delivery.
Pharmacokinetic/pharmacodynamic modelling
Surrogate parameters such as the HI viral load and the CD4 cell count provide direct markers for success or failure of HAART. Thus, diverse models correlated dose and plasma concentration of antiretrovirals to the immunological and virological therapy outcome.
Viral dynamics: A number of nonlinear mixed effects mechanismbased models were established to estimate individual unknown dynamic parameters characterizing viral dynamics during HAART.
Wu et al. [63] incorporated drug concentrations of the protease inhibitors indinavir/ritonavir, adherence and drug susceptibility into a function of treatment efficacy, defined as an inhibition rate of viral replication. Forty-four patients who failed their first protease inhibitor containing treatment were randomized to two different indinavir/ ritonavir regimens, taking either 800/200 mg BID or 400/100 mg BID. However, viral parameters identified conferring to the efficacy of HAART was the subject-specific pharmacokinetics of antiretrovirals and phenotypic drug susceptibility to HAART. It was shown that standard regression/correlation analyses could not identify significant relationships between antiviral response and drug exposure or susceptibility. Finally, Bayesian estimation approaches were able to fit viral load data for the individual subjects, such as fluctuation and viral rebound, to the model and identify the complicated pharmacodynamic relationships with confounding factors. As an example for this approach the final model of Wu et al. [63] is shown in equation 1.
Equation 1: Emax model for the drug efficacy of a single agent.
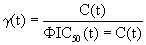 (1)
(1)
γ(t) ranges from 0 to 1 and indicates the drug efficacy; C (t)=drug plasma concentration; Φ=conversion factor between IC50(t) in vitro and IC50(t) in vivo.
However, the model employed by Wu et al. (2005) identified similar effects of the four pharmacokinetic parameters Ctrough, C12hour, Cmax and AUC on virological response, thus providing useful information for future analyses, as Ctrough is the easiest to obtain in clinical settings. Adherence, measured by pill count did not improve the pharmacodynamic model and the drug susceptibility provided instead more additional information to the adherence as the susceptibility of the virus to protease inhibitors is expected to depend on adherence over a longer period of time, which can only be estimated roughly by pill counts. Most complicated seems to be the application of mathematical models for HIV dynamics to clinical data. However these have resulted in important findings on the pathogenesis of HIV infection. A HIV viral dynamic model incorporating the effect of HAART, consisting of NRTI and PI, is a system of non-linear differential equations, as given in equation 2 [64].
Equation 2: HIV viral dynamic model incorporating the effect of an antiretroviral regimen.

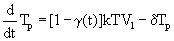
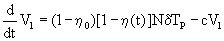
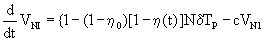 (2)
(2)
T=target uninfected cells; Tp=infected cells; V1=infectious virions; VNI=non-infectious virions; λ=rate of generation of new cells, p=proliferation rate, Tmax=T cell population density at which proliferation shuts off; dT=rate of death of uninfected cells; δ=rate of death of infected cells; k=infection rate; c=clearance of free virions; N=number of virions produced from infected cell during its life-time; η0=proportion of non-infectious virions before initiation of therapy; γ(t)=time varying Emax model as defined in 2.
This model, taken here as an example, describes non-linear functions for the number of target uninfected cells, infected cells, infectious virions and non-infectious virions, respectively. It includes the (i) rate at which new T cells are generated within the body, (ii) the T cell population density at which proliferation shuts off, (iii) the infection rate and (iv) the rates of death of infected or uninfected T cells, (vi) the number of virions produced from infected cells during their life-time, (vii) the clearance rates for free virions and the (viii) proportion of non-infectious virus in the total virus pool before initiation of therapy. Huang et al. [65] then included time-dependent parameters and drug Emax models of PI and NRTI, respectively as given in equation 3.
Equation 3: Best model and sum of squared deviations from a viral dynamic model fitting for individual subjects using the Kruskal-Wallis test and the sign test, for a protease inhibitor (indinavir/ritonavir) containing antiretroviral therapy:
A (t) = 1 and IDV/RTV Ctrough and IC50 (t),
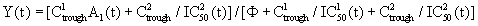 (3)
(3)
A represents the pill count at time (t), IDV/RTV=antiretroviral agents indinavir/ritonavir, Ctrough=trough plasma concentration; IC50=Concentration at which 50% of viral replication is inhibited; Φ=conversion factor between IC50(t) in vitro and IC50(t) in vivo.
Finally, a method for the determination of the inhibitory potential oh anti-HIV drugs should be discussed, although it has not found broad intention yet. Shen et al. [66] presented a work including the instantaneous inhibitory potential, IIP, of antiretrovirals into clinical consideration. The IIP includes the initial slope of the log-reduction of viral load into a median effect model, based originally on the IC50 of a drug in vitro and the measured plasma concentrations in vivo. The initial slope values have a marked effect on antiviral activity, thus complementing the information given by IC50 and IQ. The authors state, “that a drug with m=3 (m is the slope-parameter) in equation 4 produces a 10.000-fold greater inhibition at IQ=100 than a drug that would be judged equally potent based on the IC50 or IQ, but with m=1”. The authors conclude that conventional pharmacodynamic indices are insufficient to appraise the real antiviral activity of different drugs. This concept, however, has been used in drug development so far, but not in clinical considerations regarding HAART for, e.g. multiple pretreated patients and of course, prospective studies regarding this issue are lacking [66,67].
Equation 4: Slope and instantaneous inhibitory potential:
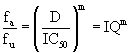 (A)
(A)
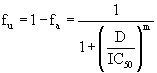 (B)
(B)
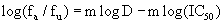 (C)
(C)
In equation (A) fa is the fraction of binding events affected or inhibited by a drug, fu is the fraction uninhibited, D is dose, IC50 is the dose causing 50% inhibition of the virus, and m is the slope parameter.
Equation (B) directly calculates the fraction of virus that are inhibited or not by a drug and equation (C) linearizes the dose-response curves by plotting log( / ) a u f f vs. log(D) ; m is the slope of this line.
Immunological response: Another approach is the modeling of the CD4 cell count evolution under protease inhibitor containing HAART and the relation to the emergence of opportunistic infections presented by Binquet et al. [68] as shown in equation 5. Immunologic response to HAART also is an important parameter of clinical efficacy, but as the CD4 cell evolution is not directly correlated to viral load decrease difficult to predict in the individual patient. However, it was shown that a rapid increase was apparent during the first two months of therapy (an average of 23.5 cells/mm³/month) subsequently slowing down the following 10 months (6.4 cells/mm³/months). After 120 days each 50 cell/mm³ increase in CD4 cell count was associated with an average 60% decrease of the incidence of opportunistic infections. However, up to date, no direct correlations between CD4 cell evolution and viral load decline could be modelled for protease inhibitor containing therapies. Recent publications suggest that HIV protease inhibitors block the apoptosis of CD4 cells independently from their antiviral efficacy [69,70].
Equation 5: Model for the CD4 cell evolution under HAART.
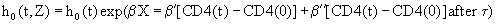
Z=vector of explanatory variables, X=vector of fixed covariates, included in Z; CD4(t)–CD4(0)=CD4 value estimated by a linear mixed effects model, measured by β ′ before time τ and by β ′ + β ′′ thereafter.
Children
Drug concentrations in children are very difficult to predict due to their developmental changes strongly affecting the bioavailability of antiretroviral drugs. Data about the pharmacokinetics of HAART in children, especially below the age of three years, are scarce. A limited number of heterogeneous studies on the use of abacavir, nevirapine, efavirenz, lopinavir [71-73], saquinavir, nelfinavir [46,49] and indinavir [74] can be found and the majority shows a very high variability of plasma concentrations in children with the potential of suboptimal drug exposure. Children therefore would be a group for an individualized dose adjustment on basis of consecutive pharmacokinetic assessments during development. Current generalized dose recommendations may not be suitable for the individual pediatric patient [72,74].
Some population pharmacokinetic approaches tried to relate children’s demographics, and changing physiological parameters to ARV pharmacokinetics, i.e. zidovudine [75], abacavir [76], nelfinavir [46,49] and enfuvirtide [57,59], and recently lopinavir [48]. It was found that especially body weight, body surface area and drug clearance changing with age are most predictive for the pharmacokinetics of ARVs.
Pregnancy
HAART during pregnancy also needs close monitoring. It has been shown in several TDM studies that plasma concentrations of most PI are substantially reduced especially in the third trimester [77]. Pathophysiological changes in absorption due to reduced gastric acid secretion, prolongued gastric and intestinal transit time, an increased volume of distribution due to an increase of body fat and water and alterations in hepatic and renal drug elimination affect at least saquinavir [78], nelfinavir [79], indinavir [79] and lopinavir, and most probably also other protease inhibitors. Data about nevirapine are contradictory and plasma concentration decreases [80] were as well reported as unchanged exposure, but higher variability [81,82]. A TDM-based dose optimization of nevirapine during late pregnancy has not been investigated yet, although recent publications showed the emergence of NNRTI resistance mutations after therapy with nevirapine in the third trimester [83].
One study with saquinavir/ritonavir 800/100 mg plus nucleosides BID defined a target plasma AUC of >10 μg×h/mL and increased the dose up to saquinavir/ritonavir 1200/100 mg BID if patients did not reach the target plasma concentrations after two weeks on treatment [78].
A successful use of TDM in pregnancy suggests pharmacokinetic assessments on time for a potential dose optimization, but current knowledge and clinical trials addressing this question are rather rare. Only one study is available, evaluating the population pharmacokinetics of nelfinavir, a protease inhibitor not used any more during for the prevention of mother-to-child transmission of HIV [48].
Ethnic differences in pk/pd of HAART
To date there is certain knowledge about ethnic differences influencing the pharmacokinetics of HAART, especially regarding genetic variations of the cytochrome P450 expression and a variety of host receptors, cytokines, chemokines, cellular and transcriptional factors. The variation of cytochrome expression in different ethnic groups with a substantial influence on plasma concentrations of antiretrovirals has been described. 15% of the asian/oceanian population are poor metabolizers of nelfinavir due to a decreased expression of CYP2C19 in comparison to only 2-4 percent of the caucasian, african, africanamerican, arabian or native australian population [84]; and 3 to 4% of caucasians are poor metabolizers of efavirenz due to a polymorphism of CYP 2B6. Although the knowledge about ethnic differences in pharmacogenomics is growing rapidly, the impact on clinical TDM is rather small. Actually, there is no recommendation for a pre-emptive screening of pharmacogenetics in patients commencing HAART and only two population-pharmacokinetic studies retrospectively evaluated the impact of ethnic variations in the pharmacokinetics of efavirenz, nevirapine, nelfinavir and indinavir [50,54,55,61].
Renal/hepatic impairment
Changes in pathophysiological states can affect hepatic or renal function und thereby change drug disposition [85-93]. Adjusting the ARV dose can be necessary in case of hepatic (protease inhibitors, NNRTI) or renal impairment (NRTI). Progression of liver damage increases the risk for markedly elevated protease inhibitor or NNRTI concentrations and it has been described that patients with replicating hepatitis B/C viruses with or without signs of hepatic impairment exhibit markedly increased drug concentrations, leading to toxic reactions [88]. Thus a replicating HBV or HCV infection is an indication for a close monitoring of HAART, and perhaps dose adjustment or as a final consequence a change of therapy, if dose adjustment is unable to decrease or avoid certain toxicities. Simulation models may help to individually adjust doses according to patient’s renal or hepatic status. However, models for this approach are lacking. TDM should also be considered in patients with chronic gastrointestinal diseases where mal absorption may occur.
Toxicities
In relation to toxicities, high plasma concentrations of protease inhibitors and NNRTI have been associated with renal/urological toxicity (indinavir) [74,94], gastrointestinal disorders (ritonavir, nelfinavir, lopinavir, saquinavir) [34,95], hyperbilirubinemia (atazanavir) [96-98], hyperlipidemia (lopinavir/ritonavir, efavirenz) [99-101] and central nervous system side effects (efavirenz [30,102]). The controversial results on elevated lipids and lipodystrophy led to the conclusion that these toxicities may be due to multifactorial geneses, including host genetics and are time-dependant, increasing the risk for body fat composition changes over cumulative time on treatment [103].
One study found that a dose reduction of indinavir in a small number of patients reduced renal urological complications, but could not find a reduction in toxicity with a dose adjustment for nelfinavir. Another observational trial observed a higher rate of CNS toxicity with efavirenz levels above the target concentration range (24%) compared to patients within the target concentration range (9%).
Drug-drug interactions
Drug-drug or drug-food interactions may result in reduced efficacy or concentration-related toxicity. All protease inhibitors and NNRTI are metabolized by cytochrome P450 isoenzymes [104], which are apparent in the intestinal mucosa and in hepatocytes [105-109], and therefore are subject to interactions among each other and with other drugs. Protease inhibitors are also substrates of a number of cellular transmembrane efflux proteins, such as P-glycoprotein and multidrug resistance proteins (MRP-1 and 2) [110-114]. These transmembrane transporters can limit the absorption of protease inhibitors and the permeation into sanctuary sites for HIV such as brain, lymphocytes, testes and macrophages. These interactions can affect drug concentrations in target tissues and plasma, and although the impact on plasma concentrations is less obvious, the variability in the admittance of antiretroviral drugs to certain compartments in vivo may have a substantial influence on therapy outcome of HAART [115]. Nevertheless, knowledge about the clinical implications and possible changes in HIV therapy as reaction to e.g. genetic deviations in one of the transporter genes remain restricted and there is no concept of personalization of HIV therapy based on results of pharmacogenetic research so far.
Much more is known about drug-drug interactions involving cytochrome P450 isoenzymes. There is a clear recommendation for TDM if a therapy regimen contains combinations of double PI, PI + NNRTI or NNRTI/PI + known enzyme inducers or inhibitors. Other key interactions affecting plasma concentrations of protease inhibitors and NNRTI are with acid reducing agents, such as proton pump inhibitors, H2-receptor blockers and antacids, furthermore anti-TB therapy or anti-neoplastics.
Several population pharmacokinetic studies evaluated the modulating effects of co-medications on HAART pharmacokinetics without adding new information to previous two-step approaches, at least regarding clinical dose recommendations. Reduced or enhanced bioavailability due to induction/inhibition of drug metabolism or reduced bioavailability due to altered absorption is no new information on drugs which have previously been extensively pharmacokinetically evaluated. Up to date, CYP or drug transporter genetics have not expanded into pk/pd modeling of drug-drug interactions.
Only a small number of interventional studies evaluated the question if the personalization of HIV therapy is superior to standard of care.
Currently, eight controlled clinical trials evaluating the role of TDM for safety and efficacy of ART have been published.
Two of these can show a significant therapeutic advantage for patients through TDM, i.e. the proportion of patients with a low viral load is higher after 48 weeks on therapy than in the control group. However these have been evaluated in trials with old PIs indinavir and nelfinavir, which are not used any more in modern ART [27,28].
Three further studies miss the goal to show a significant advantage of TDM modulated ART [29-31], One study carried out with efavirenz defined a plasma concentration cut-off for an efficient antiviral treatment of 1 μg/mL and an AUC of 60.0-120.0 μg×h/mL. Concentrations below this level were seen to confer development of viral resistance and concentrations above 4.0 μg/mL were correlated to increased CNS toxicity [30]. Efavirenz doses of patients who did not match with these criteria or experienced adverse events were altered successfully. Naturally, the reported results could not show statistically significant differences between the groups and were not sufficiently powered to proof non-inferiority of the TDM-based individual dosing. A second substudy of efavirenz treatment in 50 children successfully evaluated the use of intrapatient variability as predictor for therapy outcome [31], but although a considerable number of these children showed efavirenz AUCs below 60.0 μg×h/mL, no dose alterations were made.
At least Best et al. could show 2007 a better virological response after TDM based dose escalation of a PI [116]. A recently published work of Demeter and colleges could indeed not show a general clinical benefit towards a better virological response through TDM based dosing, but subgroup analyses showed that Hispanic and African American patients could profit and those who’s HIV showed at least a partial response to one or more of the PIs which were part of ART [117]. Also another strategy of dose escalation could only improve therapy response in patients whose virus showed at least a partial susceptibility to a PI-based ART [118].
In general, a meta-analysis of the Cochrane database of HIVTDM studies conducted between 2002 and 2007 (n=8) valuated the results critically. The methodological quality was considered good, but sample sizes being too small (n=40 to 230), too little information about randomization and also the heterogeneity of approaches, complicating a comparative meta-analysis. As a final result of this analysis it was stated by the authors that a general TDM should not be recommended in PI- or NNRTI-based ART, but the probability of virological response in patients taking an unboosted PI could be improved through TDM by 49% [119].
Also in one non-controlled trial a correlation between Ctrough of an NNRTI and virological response to ART was reported and a number of further retrospective analyses produced prediction models for therapy success and/or the emergence of adverse events, thus regarding pharmacokinetics of ARVs in patients generally as relevant for therapy success [48,49,59,61,65,120-122]. Regarding protease inhibitors, one study investigated the pharmacokinetics of lopinavir/ritonavir dosed 230/57.5 mg/m² body surface area (BSA) in children aged between birth and 12 years. In case lopinavir Ctrough were below 1 μg/mL the children received a higher dose of lopinavir 300/75 mg/m² BSA [120]. Therapeutic outcome was similar in all children. A third study assessed pharmacokinetics in 13 pregnant women receiving 800/100 mg saquinavir/ritonavir as HIV transmission prophylaxis during the third trimenon of pregnancy. If the women displayed a saquinavir AUC lower than 10.0 μg×h/mL their saquinavir dose was increased up to 1200 mg [78]. All women were successfully treated and none of the children was HIV-1 positive after birth.
Recently a French working group presented data on the TDM of efavirenz and a successful tailoring of efavirenz doses in adult outpatients. The target concentrations of 1-4 μg/mL were reached in all patients after individualizing the efavirenz dose. Unfortunately these results did not indicate whether patients had less side effects after dose alteration, or whether this group of patients was compared to a standard dosing study arm (REF: HIV Pharmacology Amsterdam 2009).
These examples show, that dose alterations were part of simple clinical study protocols and were successfully used to increase the efficacy of plasma concentration governed HAART. Nevertheless, none of these studies used population pharmacokinetics, PC-based simulations of dose or dosage interval changes and show that the integration of these still have not yet been introduced into clinical utility.
It is current consensus that TDM can be useful in the evaluation of non-compliance, drug-related toxicities, provided that target concentrations are defined for this question, and in special patients groups, were the uncertainty about deriving sufficient or non-toxic plasma concentrations is due to a lack of experience and/or data, e.g. in children or pregnant women.
Nevertheless, personalizing HIV therapy on basis of the pk-derived prediction of exposure-response relationships, e.g. of the correlation between plasma concentrations, viral resistance and viral load decline, has not yet been used in practice for a pharmacological management of antiretroviral therapy. The currently best available approaches may be the use of genotypic or phenotypic inhibitory quotients [36,38-42,121,123-127], which is nevertheless methodologically not sound up to date. A predictivity of 80-97% for therapy response for different GIQ models is satisfying from a clinical point of view, but predictivity for therapy failure still ranges somewhat between 64 and 80%, showing a considerable lack of sensitivity, which in itself already makes the applicability of such models for their clinical use arguable.
Pharmacokinetic modelling of HAART correlated to patients´ demographics is already part of drug development and pharmacological science. Pk/pd models of HAART include the effects of drug potency, pharmacokinetics, adherence, drug resistance and viral dynamics on therapy outcome. However, the number of comprehensive models is scarce, their implications for clinical therapy remain restricted.
In addition to the above mentioned, despite all efforts for the personalization of HAART, there are problems arising from the manufacturers side. There is a trend towards oral formulations of ARVs which are dosed higher than previously in order to reduce the pill burden for patients and once-daily therapy regimens. Although this unquestionably contributes to patient´s compliance and quality of life, it deprives clinicians to tailor ARV doses individually due to the patient´s individual demands. The earlier change from saquinavirmesylate formulations of 200 mg per capsule to 500 mg per tablet, the increase of lopinavir/ritonavir dose from 133/33 mg per capsule to 200/500 mg per film-coated tablet foils the efforts to personalize HIV therapy and the reduction of a dosage in order to reduce apparent toxicities bear a higher risk of losing therapeutic efficacy. Only lately lopinavir/ritonavir has been approved as 100/25 mg tablet formulation, which can be used now for individualizing doses to patients. Fixed dose combinations forge ahead on the market, Atripla® (efavirenz/tenofovir- DF/emtricitabine) today has strong comparators such as Complera (rilpivirine/tenofovir-DF/emtricitabine)® and Stribild® (elvitegravir/ cobicistat/tenofovir-DF/emtricitabine) and also the INI dolutegravir, which probably will be marketed together with another fixed dose combination Kivexa® (lamivudine and abacavir) [128].
In general, dose alterations of standard NNRTI regimens should be applied very carefully, due to of the very low resistance barrier. Still, body weight derived dose adaption is useful, e.g. in case of pediatric HIV therapy or patients with a very low or very high body weight. And it certainly is useful in order to estimate assumed drugdrug interactions, e.g. with tuberculosis comedication, or metabolism disorders in case of e.g. actively replicating hepatitis B/C virus with signs of hepatic impairment.
Personalizing ART certainly is an issue in lifelong therapy, troubled with side-effects and pill burden, given to millions of individuals worldwide with different ethnic backgrounds and characteristics. Ways to apply personalized therapy to clinical use have been shown by mathematicians, clinicians, biologists. However, the reality shows that prospective studies regarding this issue are still lacking, that the trend towards single tablet regimes and fixed dose combinations are foiling such efforts. Today, there are more antiretroviral drugs and classes on the marked than ever, more insight into mechanisms of viral inhibition and effect models, but ways to individualize therapy have become less.