Journal of Nanomedicine & Biotherapeutic Discovery
Open Access
ISSN: 2155-983X
![]() +44 1300 500008
+44 1300 500008
ISSN: 2155-983X
![]() +44 1300 500008
+44 1300 500008
Research Article - (2016) Volume 6, Issue 1
Quantum Dots
(QDs) are rapidly becoming popular as novel tools for theragnostic purposes. This report evaluates the pharmacokinetics parameters of CdS-MDx QDs of different tissues following a single dose i.p. to rodents at several time points, as a model system for determining their tissue uptake, time of residence and elimination. We employed an analysis of tissue images using fluorescence microscopy and tissue homogenates by spectroscopy to identify and measure the CdS-MDx QDs content and analyze the concentration of QDs in each tissue at predetermined time intervals. The pharmacokinetics analysis of CdS-MDx QDs (Cmax, Tmax, AUC0-t, AUC0-∞, Ke and MRT) were different for each tissue after a single dose of QDs during 360 h. Liver and
kidney tissues
took up the most QDs, but their Ke and MRT evidenced a rapid kinetic of elimination, suggesting QDs might get eliminated by these organs. Our data clearly showed that CdS-MDx QDs were not completely cleared from in vivo systems after 360 h. CdS-MDx QDs appears to be nanomaterials with favorable pharmacokinetics properties to develop novel therapeutic and diagnostic modalities.
Keywords: Quantum dots; Pharmacokinetics; Area under curve; Cadmium sulfide; Elimination; Constant of elimination; Mean resident time
QDs: Quantum Dots; CdS-MDx: Maltodextrin Capped Cadmium Sulfide; Cmax: Maximum Concentration; Tmax: Time of Maximum Concentration; Ke: Elimination Constant; AUC0-t: Area under Concentration Time Curve from Zero Hours to Time; AUC0-∞: Area under Concentration Time Curve from Zero Hours to Infinity; MRT: Mean Residence Time
Nowadays, Quantum Dots (QDs) are revolutionizing biomedical science not only by increasing the understanding pathological basis of diseases, by also offering new tools for theranostic purposes [1]. Although the new technologies are facilitating the study of QDs interactions with the bio phase, through monitoring of fate of QDs in organism, there are a few studies focused to study the pharmacokinetics parameters of these nanomaterials [2]. Pharmacokinetic studies the movement of drugs in the body and reveals its concentration, depending on dose and time elapsed since its administration. The pharmacokinetic of a conventional drug and thes nanoparticles are often different because of the characteristics of nanomaterials, such as particle size, surface area, surface chemistry, chemical composition, encapsulation, conjugated molecules and biocompatibility [3-5]. A primary challenge of nanotechnology is to synthesize nanomaterial with characteristics that allow it to become available in a useful concentration in the target tissue after injection into living subjects [6].
Currently, all available reports have unanimously concluded that QDs show a preference for deposition in organs and tissues and that they do not remain circulating in the bloodstream, which is an advantage if we want to deliver a drug-containing QDs to specific tissues [7,8]. The pharmacokinetic parameters of the behavior of cadmium-containing QDs in biological systems are still being researched. However, expert reports point out that the biological behavior of cadmium-containing QDs, including the kinetics of uptake, transport, and clearance in the body and target organs needs to be characterized in order to understand and predict their efficacy and safety [3,9,10].
We recently synthesized maltodextrin capped Cadmium Sulfide QDs (CdS-MDx) [11,12]. Although the in vitro and in vivo studies revealed that these CdS-MDx QDs produced distinct dose-dependent toxic effects, the in vivo studies demonstrated that, when administered to rodents, CdS-MDx QDs were biocompatible and non-toxic after 5 and 15 days. Nevertheless, the pattern of biodistribution and accumulation of CdS-MDx QDs was different in the tissues after repeated doses [13]. Therefore, this paper analyze a series of pharmacokinetics parameters of CdS-MDx QDs, such as Cmax, Tmax, AUC0-t, AUC0-∞, Ke and MRT of different tissues after a single dose i.p. to rodents at several time points, in an attempt to offer new insights into the uptake, time of residence and elimination of CdS-MDx QDs in specific organs.
Analysis of kinetics of tissue bio distribution of CdS-MDx QDs in rats
CdS-MDx QDs with a diameter of about 3.5 nm were synthesized as previously described [11]. The Wistar rat was selected as the model for the pharmacokinetic study of CdS-MDx QDs. All animals were kept in hygienic and animal-friendly conditions, housed in a temperature and humidity controlled environment, and allowed food (Standard Purina Chow Diet, Mexico) and water ad libitum. The experiments were conducted in accordance with the Guide for the Care and Use for Laboratory Animals [14].
Healthy male rats (8-10 weeks old) were selected randomly and divided into two groups: in one group, 6 animals were treated with a single i.p. dose of PBS and were used as control; in the second group, sixty animals were treated with a single dose of CdS-MDx QDs (200 μg/Kg) body weight i.p. CdS-MDx QDs was dissolved in 300 μL PBS. Dose chosen was based on previous studies on biocompatibility of CdS-MDx QDs in rats performed at 5 and 15 days, where was revealed the distribution pattern of QDs and their biocompatibility with all tissues analyzed [13]. Animals were sacrificed at 0, 7, 10, 18, 24, 48, 72, 144, 216, 288 and 360 h. During the study, animals were observed for signs of toxicity. Signs recorded during acute toxicity included: motor activity, anesthesia, tremor, arching, and rolling, clonic seizures, ptosis, tonic extension, lacrimation, exophthalmos, pilo-erection, salivation, depression, ataxia, sedation, hypnosis, cyanosis and analgesia. Behavior parameters, death, the weight, and the amount of water and feed were analyzed. Toxicity indices consisted of daily clinical observations, body weight, food consumption, clinical pathology, organ weights, and histopathology.
After treatment, animals were fasted overnight and blood samples were obtained from the heart following anesthesia with ether. Tissues collected at necropsy were preserved in 10% neutral-buffered formalin fixative and were processed for routine histologic examination. In order to obtain precise images regarding the tissue distribution of the QDs, unstained tissue samples were analyzed under a fluorescence microscope. In order to reduce auto-fluorescence, tissues were rinsed many times with 1 mg/ml solution of sodium borohydride, as blocking agent. For the pharmacokinetic profile of CdS-MDx QDs, we performed a quantitative analysis of fluorescence in tissue samples from liver, kidney, lung, intestine, heart, spleen, brain, and testis. For this, 50 mg of tissue were homogenized with 1 mL of lysis buffer (20 mM HEPES, 2 mM EGTA, 50 mM β-glycerol phosphate, 5 mM sodium fluoride, 50 μM Dithiothreitol (DTT), 100 mM Phenylmethylsulfonyl Fluoride (PMSF)). The intensity of fluorescence was measured in a spectrophotometer (Perkin Elmer) using the excitation wavelength of 485 nm.
The analyzed pharmacokinetic parameters included the maximum observed tissue concentration, (Cmax), time to reach Cmax, (Tmax), area under the concentration-time curve (AUC) from time zero to the last time (AUC0-t), the AUC from time zero extrapolated to infinity (AUC0-∞), the elimination constant (Ke) and Mean Residence Time (MRT) according the next equations:
Ke =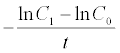
AUC0-t=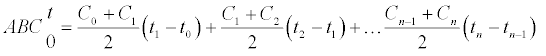
AUC0-∞= 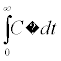
MRT = 1 / Ke
Maximal Concentration (Cmax) was considered as maximal concentration achieved by QDs. Time of Maximum Concentration (Tmax) was considered as the time at which Cmax was achieved by QDs.
Statistical analysis
The data were represented as the mean ± SD. The data was statistically analyzed using the SPSS 10.0 software (SPSS Inc., Chicago, Ill., USA), the t-test, and ANOVA. Differences were considered significant if the P-value was less than 0.05.
In order to obtain precise data regarding the kinetic of tissue biodistribution of CdS-MDx QDs, we quantified the amount of CdSMDx QDs present in tissues using spectrophotometry and visualized them in unstained tissue samples under a fluorescence microscope. CdS-MDx QDs were identified because of their bright green light.
Pharmacokinetic parameters derived from quantitative analysis of the i.p. administration of CdS-MDx QDs are summarized in Table 1.
| Tissue | Cmax (UA) | Tmax (h) | AUC0-t (UA * h) | AUC0-8 (UA * h) | Ke (h - 1) | MRT (h) |
|---|---|---|---|---|---|---|
| Liver | 15,275 | 72 | 3,908,265 | 3,938,883 | 0.0108 | 92 |
| Kidney | 11,723 | 24 | 1,569,306 | 1,595,387 | 0.0103 | 97 |
| Heart | 9,029 | 72 | 2,324,674 | 2,827,714 | 0.0051 | 196 |
| Lung | 4,424 | 10 | 517,734 | 768,566 | 0.0035 | 285 |
| Intestine | 9,009 | 48 | 2,438,770 | 5,300,398 | 0.0017 | 588 |
| Spleen | 4,328 | 72 | 995,710 | 2,086,957 | 0.0020 | 500 |
| Brain | 3,740 | 72 | 1,096,270 | 3,580,898 | 0.0011 | 909 |
| Testis | 3,048 | 48 | 618,496.8 | 845,755.6 | 0.0033 | 303 |
Table 1: Pharmacokinetics of CdS-maltodextrin QDs. Cmax: Maximum Concentration; Tmax: Time of Maximum Concentration; AUC0-t: Area under Concentration Time Curve from Zero Hours to Time; AUC0-8: Area under Concentration Time Curve from Zero Hours to Infinity; Ke: Elimination Constant.
Mean tissue concentrations of CdS-MDx QDs in rats are shown in Figure 1.

Figure 1: Tissue concentrations (mean ± SD, n = 6) of CdS-MDx QDs following intraperitoneal administration at one dose rate of 200 μg/Kg to clinically healthy rats.
Tissue concentration peaks of CdS-MDx QDs were reached at different times (tmax): at 10 h in lung, at 24 h in kidney, at 48 h in intestine and testis, and at 72 h in liver, heart, spleen and brain. Following an i.p. dose, however, CdS-MDx QDs demonstrated a significant high AUC0-t. This happened in higher liver, intestine, heart, and kidney. The mean Cmax values were higher in liver, kidney, heart and intestine (15275, 11723, 9,029 and 9009 UA, respectively). The Ke of CdS-MDx QDs was higher in liver and kidney (0.0108 and 0.0103 h - 1, respectively) and lower in intestine and brain (0.0017 and 0.0011 h - 1, respectively). That results were according with the Mean Residence Time values (MRT; i.e., the average time that the QDs stay inside the tissue) of CdS-MDx QDs. We found a short MRT in liver and kidney, and a very large one in brain, intestine and spleen (909, 588 and 500 h, respectively). Present results show that CdS-MDx QDs have different bio distribution kinetics in different tissues. They reach high concentrations in tissues such as liver, intestine, heart, and kidney, but their rate of elimination was different. The liver and kidney almost completely eliminated the CdS-MDx QDs, while the brain did so much more slowly.
Figures 2 and 3 show representative fluorescence images of the biodistribution of CdS-MDx QDs in liver, kidney, heart, lung, intestine, spleen, brain, and testis from rats at 7 h, at time of Cmax, and at 360 h. The visualization of differing fluorescence intensity due to the presence of CdS-MDx QDs in each tissue was evident since 7 h. Such intensity was visualized at different times (10 h – 72 h) in studied tissues, and at the end of the study (360 h). A histological analysis of stained tissue was also undertaken in order to monitor their toxicity and see if CdS-MDx QDs were biocompatible or if they induced structural changes. Non-morphological changes were observed at 360 h.

Figure 2: Fluorescence microscopy images showing the distribution and localization of CdS-MDx QDs in tissues from rats. The images belong to the liver, kidney, heart, intestine, and stomach from male rats treated with a single dose of 200 μg/Kg of QDs. Tissue samples were analyzed under fluorescence microscopy at 20X. Column A shows tissues at 7 h, the first time of analysis; column B shows images of tissues observed at the time of maximal concentration (Tmax); column C shows images of tissues at 360 h; column D shows tissues stained with H&E at 360 h. The distribution and localization of CdS-MDx QDs was identified by a bright green imaging in the analyzed tissues.

Figure 3: Fluorescence microscopy images showing the distribution and localization of CdS-MDx QDs in tissues from rats. The images belong to brain and testis from male rats treated with a single dose of 200 μg/Kg of QDs. Tissue samples were analyzed under fluorescence microscopy at 20X. Column A shows tissues at 7 h, the first time of analysis; column B shows images of tissues observed at the time of maximal concentration (Tmax); column C shows images of tissues at 360 h; column D shows tissues stained with H&E at 360 h. The distribution and localization of CdS-MDx QDs was identified by a bright green imaging in the analyzed tissues.
The PK of nanoparticles is determined by a number of interrelated physicochemical and biological factors, and in-depth understanding of the particle characteristics (both before and after injection into living subjects) is critical for selecting QDs and will provide important guidelines for their successful clinical use [2,15]. It has been previously shown that QDs are distributed to multiple organs. However, there is no consistent data about the time that they remain in the body and current reports point out the need for further studies addressing the biological behavior of QDs, including the kinetics of uptake, transport, and clearance in the body. In this study, it was monitored the kinetics of tissue bio distribution after a single i.p. dose of CdS-MDx QDs at several time points in rats in order to establish the amount of uptake by tissues, the mean time of residence, and the rate of elimination.
Some studies on the pharmacokinetics of cadmium-containing QDs have been carried out, but they are not comparable because the dosage, route of entry into the body and the time of monitoring are different [16,17]. QDs, when administered by intravenous injection, have different behaviors. It has been found that, one hour after intravenous injection; all CdSe QDs had concentrated inside the spleen, liver, and kidney, while no QDs were found in others tissues [18]. Kato et al. [19] demonstrated that the liver takes up 40% of QD-BSA after 90 min and those QDs can remain up to ten days after intravenous dosing. Another study using the intravenous route demonstrated that CdTe / ZnS QDs revealed an accumulation in the liver, kidneys, and spleen and authors concluded that QDs could undergo degradation in vivo [20].
The intraperitoneal route is commonly used in preclinical studies for administering drugs because this pathway reaches systemic blood circulation. This study assessed whether CdS-MDx QDs administered intraperitoneally in a single dose were delivered into various organs in an in vivo rat model. It has been demonstrated that QDs administered intraperitoneally were delivered via systemic blood circulation into liver, spleen, kidney and brain at 6 h, and it was also demonstrated that QDs were still circulating throughout the body at 6 h after injection [21]. We found that 7 h after the administration CdS-MDx QDs, these could already be found in various organs / tissues, including liver, kidney, intestine, lung, heart, spleen, brain and testis, and were still present 360 h after administration. The time to reach the maximal concentration (Tmax) was between 24 h and 72 h; however, the presence of QDs in all tissues was evident at 7 h.
According with present results, the amount of CdS-MDx QDs remaining in each tissue was different during 360 h, as demonstrated by the AUCs. The highest accumulation was found in liver, intestine, heart, spleen and brain (AUC0-∞). However, the rate of elimination (Ke) was faster in liver and kidney than other tissues; even the Mean Time of Residence (MRT) was lower in these organs, which might suggest that CdS-MDx QDs are eliminated through said organs. A few studies have looked at the excretion of QDs following their in vivo administration, but the reports are not conclusive. Tang et al. [22] reported that QDs coated with cationic Polydiallyldimethylammonium chloride (PDDA) preferentially deposit in the lung other than in the liver, while the negative and PEGylated QDs render abundant accumulation in the liver and cause injuries in specific tissues. The kidney is regarded as the key organ for the clearance of ultrasmall NPs (< 5.5 nm). But, recent studies demonstrated that QDs in this size range could accumulate in the kidney for extended times without urinary excretion [23]. Interestingly, the present study found earlier Tmax in kidney and in liver; this might indicate that CdS-MDx QDs are quickly depurated by these organs. It has been previously reported that free QDs, which kept their original nanosize (5.5 nm), could be filtered by glomeruli and were efficiently excreted via urine [24]. Our data clearly showed that CdS-MDx QDs of 3.5 nm, were present in liver and kidney in high concentrations and their rate of elimination was faster than in other tissues; the amount of CdS-MDx QDs was almost insignificant at 360 h in those tissues. The presence of a high AUC in intestine might also indicate a biliary elimination of QDs. At 360 h, a rapid kinetic of elimination in all tissues, except brain, was evident.
The biodistribution of NPs is also determined by biological barriers, including the Blood-Brain Barrier (BBB) and Blood-Testis Barrier (BTB) [25]. Previous studies on the biodistribution of nanoparticles demonstrated their presence in the brain following intraperitoneal injection in rodents [17,26], even it has been demonstrated that QDs functionalized with a zwitterionic compact ligand can reach chick embryo brain without any apparent toxicity [27]. This group has demonstrated that CdS-MDx QDs are distributed into the brain after repeated doses in rodents and no morphological changes were observed [13]. Present results demonstrate that CdS-MDx QDs did not reach high concentrations in this organ. However, the pharmacokinetic analysis showed a very low Ke and a high MRT, which means that CdS-MDx QDs could remain in the brain for a very long period of time. Recent studies have demonstrated the neurotoxicity of QDs in the central nervous system and impairments of synaptic transmission and plasticity, as well as brain functions, in tested animals [28,29]. Further studies are needed to assess the potential toxic of CdS-Mdx QDs in brain.
The Blood-Testis Barrier (BTB) is also one of the tightest bloodtissue barriers in the mammalian body. It divides the seminiferous epithelium into the basal and the apical (adluminal) compartments and provides a physical barrier to segregate the events of spermatogenesis [30]. Previous reports by this group have also demonstrated the presence of CdS-MDx QDs in the BTB adjacent seminiferous tubules. That said, the BTB was found to be intact and functional after a single dose as well as repeated doses in rodents. A histopatological analysis demonstrated that the presence of CdSMDxQDs in testis did not produce any morphological alterations. According with previous results the small size (3.5 nm) of CdS-MDx QDs seems allows them to enter the body by crossing several barriers, to pass into the blood stream from where they can reach organs and tissues and interact with biological structures. CdS-MDx QDs, appear to stay in body for a long period of time, in some organs. Previous studies by this group have shown that when CdS-MDx QDs were administered daily during 15 days did not produce damaging their normal functions and morphology, so the specific properties that possess those QDs appear to be minimizing or eliminating their nanotoxicity.
In conclusion, we have researched the kinetics and behavior of CdSMDx QDs administered via intraperitoneal injection in a rat model. Our findings revealed the persistent nature of CdS-MDx QDs with regard to tissue kinetics. The pattern of biodistribution and accumulation of CdS-MDx QDs was different for each tissue after a single dose of QDs during 360 h. The liver and kidney showed the most uptakes, but their Ke and MRT evidenced a rapid kinetic of elimination; suggesting QDs might get eliminated by these organs. Other tissues took up lesser amounts of CdS-MDx QDs and the kinetic of elimination was slower. Our data clearly show that CdS-MDx QDs not were completely cleared from in vivo systems after 360 h. CdSMDx QDs appears to be nanomaterials with favorable pharmacokinetics properties to develop novel therapeutic and diagnostic modalities.