Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2010) Volume 3, Issue 1
Protein Tyrosine Phosphatase 1B (PTP-1B) is one of the important targets in the treatment of diabetes and obesity. They play a very important role in cellular signaling within and between cells. The best pharmacophore hypothesis (Hypo 1), consisting of four features, namely, one hydrogen- bond acceptor (HBA), one hydrophobic point (HY), and two ring aromatics (RA), has a correlation coefficient of 0.961, a root mean square deviation (RMSD) of 0.885, and a cost difference of 62.436, suggesting that a highly predictive pharmacophore model was successfully obtained. A chemical feature based pharmacophore model has been generated from known PTP-1B inhibitors (25 training set compounds) by HypoGen module implemented in CATALYST software. The top ranked hypothesis (Hypo1) contained four chemical feature types such as hydrogen-bond acceptor (HA), hydrophobic aromatic (HY), and two ring aromatic (RA) features. Hypo1 was further validated by 125 test set molecules giving a correlation coefficient of 0.905 between experimental and estimated activity. This was also validated using CatScramble method. Thus, the Hypo1 was exploited for searching new lead compounds over chemical compounds in Medichem database and then the selected compounds were screened based on restriction estimated activity. Finally, we obtained 30 new lead candidates and the one best highly active compound structure was selected as a lead compound. The results demonstrate that hypothesis derived in this study could be considered to be a useful and reliable tool in identifying structurally diverse compounds with desired biological activity.
Keywords: Protein tyrosine phosphatase 1B; Diabetes; Insulin; Obesity; Catalyst; Pharmacophore
Protein tyrosine phosphatases (PTPs) constitute a family of receptor-like and cytoplasmic enzymes that catalyze the dephosphorylation of phosphotyrosine residues in protein substrates. PTPs together with protein tyrosine kinases (PTKs) play critical roles in regulating intracellular signal transduction pathways responsible for controlling cell growth, differentiation, motility, and metabolism.
Protein tyrosine phosphatases (PTPs) have emerged as a new and promising class of signaling targets, since the discovery of PTP-1B as a major drug target for diabetes and obesity. Biochemical and cellular studies have provided evidences that PTPs have an important role in the regulation of insulin signal transduction. Protein tyrosine phosphates 1B (PTP-1B), a cytosolic PTP play a major role in the regulation of insulin sensitivity and dephosphorylation of the insulin receptor. PTP-1B has been implicated as negative regulator of insulin receptor signaling Reference (Zhang and Zhang, 2007).
Clinical studies have found a correlation between insulin resistance states and levels of PTP-1B expression in muscle and adipose tissue, suggesting that PTP-1B has a major role in the insulin resistance associated with obesity and NIDDM. Blocking one or more phosphatases could enhance the phosphorylation state of the insulin receptor kinase/subunit and/or its downstream signaling partners and restore the insulin resistance, which is a characteristic of type II diabetes (van Huijsduijnen et al., 2004).
Since then, many drugs have been synthesized by various companies for targeting PTP-1B, which is very challenging due to the closed form of the catalytic site of PTPs containing a highly polar phosphotyrosine (pTyr) binding site. The quest for oral PTP-1B inhibitors, with a satisfactory balance between physicochemical properties and selectivity, is still in its early stages, but despite the recent progress, compounds with optimal oral activity remain to be discovered. A pharmacophore model represents the 3D arrangements of the structural or chemical features of a drug (small organic compounds, peptides, peptidomimetics, etc.) that may be essential for interacting with the protein for optimum binding.
These pharmacophore models can be used differently in drug design programs such as
(i)3D query tool for virtual screening to identify potential new compounds from 3D databases of “drug-like” molecules that have patentable structures different from those that currently exist, and
(ii)Tool to predict the activities of a set of new compounds that remain to be synthesized Reference (Bharatham et al., 2007).
In the present study, we have generated pharmacophore model using Catalyst software Reference (Catalyst 4.11, Accelrys, Inc., San Diego, CA, 2005) for diverse set of molecules of PTP IB with an aim to obtain Pharmacophore model which could provide a rational hypothetical picture of the primary chemical features responsible for activity.
This is expected to provide useful knowledge for developing new potentially active candidates targeting the PTP-1B which can be useful for treatment of obesi ty and diabetes. Pharmacophore modeling correlates activities with the spatial arrangement of various chemical features.
Selection of molecules
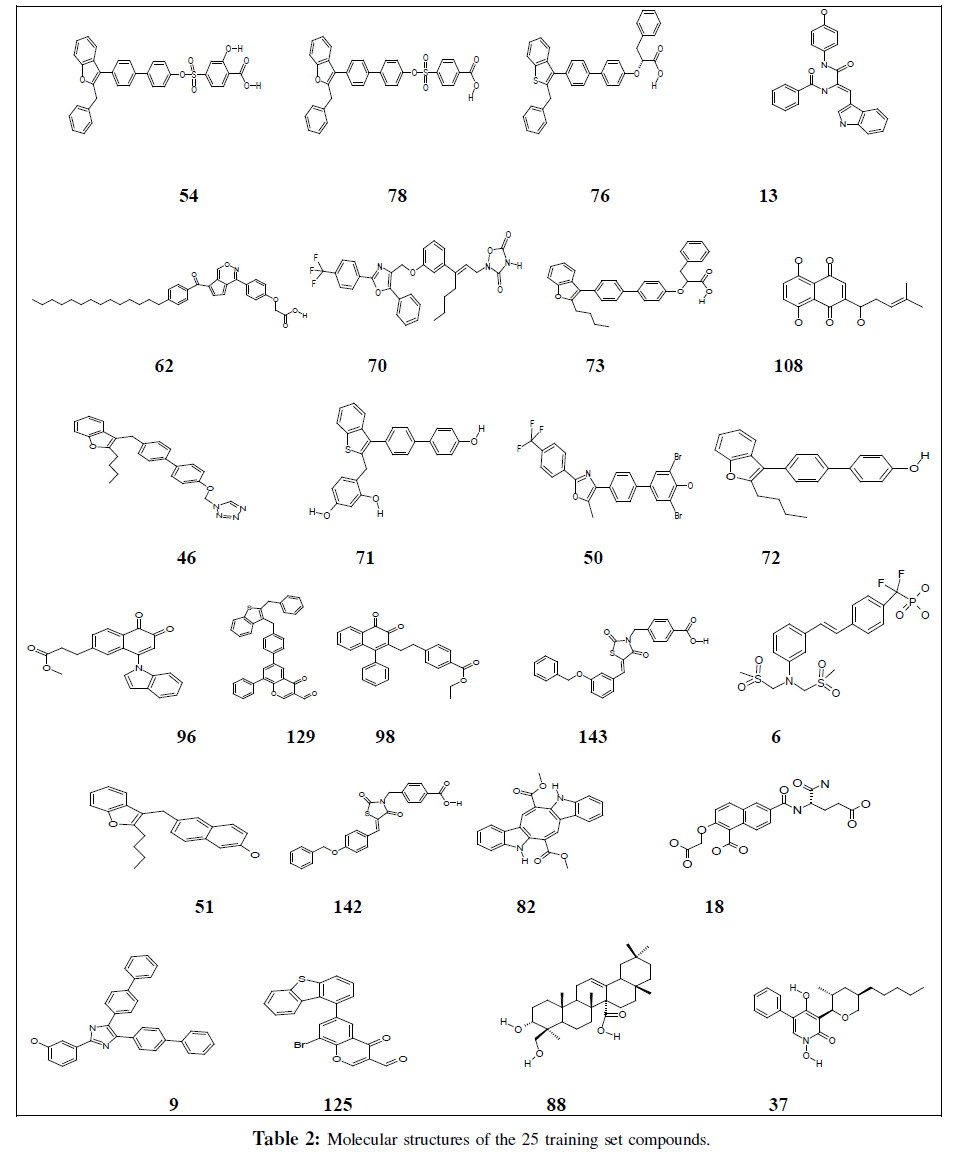
We selected a set of 150 compounds which are reported to be inhibitors of PTP-1B. The inhibitory activity of these compounds was expressed as IC50 (i.e., concentration of compound required to inhibit 50% of PTP-1B was taken). The activity reported for these compounds was measured according to assay procedures. The IC50 values spanned across a wide range from 0.039 uM to 1800 uM. Of these 150 compounds, 25 compounds were taken as training set (Table 2) and the rest of the 125 compounds as test set (Chart 2 Table S1 Additional information). The dataset was divided into training set and test set. The training set was selected by considering both structural diversity and wide coverage of the activity range. They were distributed into most active, moderately active and least inactive compounds based on their IC50 values in order to obtain critical information on pharmacophore requirements. The important aspect of this selection was to ensure that each active compound would teach something new to the HypoGen module thus it can be able to uncover as much as critical information possible for predicting biological activity.
| Compound | True IC50 (uM) | Estimated IC50 (uM) | Error factor (a) | Fit Value (b) | Activity Scale (c) | Estimated activity scale |
|---|---|---|---|---|---|---|
| 54 | 0.039 | 0.064 | 1.6 | 12.45 | +++ | +++ |
| 78 | 0.075 | 0.054 | -1.4 | 11.32 | +++ | +++ |
| 76 | 0.095 | 0.05 | -1.9 | 11.31 | +++ | +++ |
| 62 | 0.15 | 0.74 | 4.9 | 11.58 | +++ | +++ |
| 70 | 0.37 | 0.65 | 1.8 | 11.66 | +++ | +++ |
| 73 | 0.44 | 0.37 | -1.2 | 11.93 | +++ | +++ |
| 46 | 0.51 | 0.51 | -1 | 11.55 | +++ | +++ |
| 71 | 0.58 | 0.35 | -1.6 | 11.42 | +++ | +++ |
| 50 | 0.65 | 1.4 | 2.2 | 11.45 | +++ | +++ |
| 72 96 129 98 143 51 142 82 9 125 88 37 13 108 6 18 |
0.74 0.92 1 1.01 1.1 1.3 1.6 3.77 8.6 10 11.7 12 21.5 25 57.9 3800 |
0.36 0.88 0.46 0.51 3.5 1.2 1.2 2.5 1.4 25 25 6 25 25 25 1800 |
-2 -1.1 -2.2 -2 3.2 -1.1 -1.4 -1.5 -1 2.5 2.1 -2 1.1 -1 -2.4 -2.1 |
11.34 10.69 11.66 10.76 10.80 10.65 9.92 9.67 8.92 8.57 8.95 9.28 9.58 8.58 8.93 8.46 |
+++ ++ ++ ++ ++ ++ ++ ++ + + + + + + + + |
+++ ++ ++ ++ ++ ++ ++ ++ + + + + + + + + |
aThe error factor is computed as the ratio of the measured activity to the activity estimated by the hypothesis or the inverse if estimated is greaterthan measured
bFit value indicates how well the features in the pharmacophore overlap the chemical features in the molecule.
cActivity scale: +++, <0.5 uM (highly active); ++, >0.5-10 uM (moderately active); +, >10 uM (inactive).
Table 1: Output of the score hypothesis process on the training set.
Molecular modeling
The geometry of a compound is built with the Catalyst builder and optimized by the CHARMM like force field. All molecules were built using the builder module of Cerius2. All the structures were minimized using steepest descent algorithm with a convergence gradient value of 0.001 kcal/mol. Partial atomic charges were calculated using Gasteiger method Reference (Gasteiger and Marsilli, 1980). Further geometry optimization was carried out for each compound with the MOPAC 6 package using the semi-empirical AM1 Hamiltonian.
Pharmacophore modeling
Multiple acceptable conformations were generated for all of 20.0 kcal/mol above the global energy minimum. Instead of using lowest energy conformation of each compound, multiple acceptable conformations were generated for all ligands within the Catalyst ConFirm module using the ‘‘Poling’’ algorithm. A maximum of 250 conformations were generated for each molecule within an energy threshold all conformational models for each molecule in training set were used in Catalyst for pharmacophore hypothesis generation.
The training set molecules (25) associated with their conformations were submitted to the Catalyst hypothesis generation (HypoGen) (Table 1). Features of hydrogen-bond acceptor (HBA), hydrophobic features (HY), hydrogen-bond donor (HBD), ring aromatic (RA) features were included for the pharmacophore generation on the basis of common features present in the study molecules. The statistical parameters like cost values determine the significance of the model. Ten pharmacophore models with significant statistical parameters were generated.
The best model was selected on the basis of a high correlation coefficient (r), lowest total cost, and RMSD values. The final model was further validated by a test set of 125 molecules.
Generation of pharmacophore model
Based on the structures of the training set compounds and their experimentally determined inhibitory activities against PTP-1B, 10 best pharmacophore (or hypotheses) were generated using HypoGen module implemented in Catalyst 4.11 software. On further analysis, it was observed that four chemical feature types such as hydrogen-bond acceptor (HA), hydrophobic aromatic (HY), and two ring aromatic (RA) features could effectively map all critical chemical features of all molecules in the training and test sets. These features were further selected and used to build a series of hypotheses using the HypoGen module in Catalyst using default uncertainty value 3 (defined by Catalyst software as the measured value being within three times higher or three times lower of the true value). Catalyst thereby generates a chemical feature based model on the basis of the most active compounds.
In hypothesis generation, the structure and activity correlations in the training set were examined. HypoGen identifies those features that were common to the active compounds but excluded from the inactive compounds within conformationally allowable regions of space. It also further estimates the activity of each training set compound using regression parameters. These parameters are computed by the regression analysis using the relationship of geometric fit value versus the negative logarithm of activity. The greater is the geometric fit, greater would be the activity prediction of the compound.
The fit function not only checks the feature is mapped or not but whether it contains a distance term, which measures the distance that separates the feature on the molecule from the centroid of the hypothesis feature. Both these terms are used to calculate the geometric fit value.
Pharmacophore validation
The generated pharmacophore model should be able to also predict the activity of the molecules accurately and also identify the active compound from the database. Therefore, the derived pharmacophore map was validated using (i) cost analysis, (ii) test set prediction and (iii) Fisher’s test.
Cost analysis
The HypoGen module in Catalyst performs two important theoretical cost calculations determining the success of any pharmacophore hypothesis. One is the ‘fixed cost’ (termed as ideal cost), representing the simplest model that fits all data perfectly, and the second is the ‘null cost’ (termed as no correlation cost), representing the highest cost of a pharmacophore with no features and estimates activity to be the average of the activity data of the training set molecules.
A meaningful pharmacophore hypothesis may also result when the difference between null and fixed cost value is large; with values of 40-60 bits for a pharmacophore hypothesis may indicate that it has 75-90% probability of correlating the data (Catalyst 4.11 documentation).
Two other parameters determine the qual ity of any pharmacophore configuration cost or entropy cost depending on the complexity of the pharmacophore hypothesis space and should have a value <17, and the error cost, which is dependent on the root mean square differences between the estimated and the actual activities of the training set molecules. The RMSD represents the quality of the correlation between the estimated and the actual activity data. The best pharmacophore model has highest cost difference, lowest RMSD and best correlation coefficient.
Test set activity prediction
In addition to the estimation of activity of the 25 training set molecules, the pharmacophore model should also be able to estimate the activity of new compounds. For external validation of the pharmacophore model, we have considered 125 compounds as test set (Table S1 Supporting information), having wide range of activities (IC50, spanning from 0.5 to 10.00 uM) and structural diversity. The best pharmacophore (Hypo1) having high correlation coefficient (r), lowest total cost, and lower RMSD value was chosen to estimate the activity of test set. Test set compounds were classified on the basis of their activity as highly active < 0.5 uM (highly active); ++, > 0.5-10 uM (moderately active); +, > 10 uM (inactive) (Doman et al., 2002; Lazo et al., 2001; Malamas et al., 2000a; Malamas et al., 2000b; Jia et al., 2001; Gao et al., 2001; Ripka, 2000; Lyon et al., 2002; Furstner et al., 2004; Taha and AlDamen, 2005; Cho et al., 2006; Cui et al., 2006; Dewang et al., 2005; Lazo et al., 2006; Mao et al., 2006; Na et al., 2006a; Na et al., 2006b; Wang et al., 1998; Liu et al., 2003; Wipf et al., 2001; Ahn et al., 2002; Chen et al., 2002; Shrestha et al., 2004; Black et al., 2005; Leung et al., 2002; Shim et al., 2005; Cao et al., 2005; Huang et al., 2003; Maccari et al., 2007; Wrobel et al., 1999).
Fisher’s test
Using the module CatScramble, the molecular spreadsheets of our training set were modified by arbitrary scrambling of the affinity data for all compounds. These randomized spreadsheets yield hypotheses without statistical significance; otherwise, the original model is also random. To achieve a statistical significance level of 98%, 41 random spreadsheets were generated for each of our three hypotheses. For all three targets, randomization tests gave hypotheses with total cost values lying well above those reported for the sets of original hypotheses, yielding lower values for the differences null hypothesis cost - total cost, further supporting the statistical significance of our models Rituparna Sarma et al., 2008.
Pharmacophore models were generated by HypoGen present in (Catalyst 4.11, Accelrys, Inc., San Diego, CA, 2005) and top 10 hypotheses (Table 1) were exported. Most hypotheses showed high correlation (>0.90). Interestingly, in the training set, all highly active compounds map all the features that is hydrophobic (HY), hydrogen-bond acceptor (HBA), and two ring aromatics (RA1 and RA2). With a few exceptions, in moderately active and inactive compounds one feature is missing. All the compounds in the training set map HY and RA1 feature revealing that these two features should be mainly responsible for the high molecular bioactivity, thus, should be taken into account in discovering or designing novel PTP-1B. The most active compound, 54, has a fitness score of 12.45 when mapped to Hypo 1 (Figure 1) whereas the least active, 18, maps to a value of 8.46 as seen in Figure 2B (1). On the basis of similar composition of the 10 hypotheses, hypothesis 1 (Hypo1), characterized by the best statistical parameters (Table 1) in terms of its predictive ability, as indicated by the highest correlation coefficient and lowest RMS deviations, has been chosen to represent ‘the pharmacophore model’. Remarkably, the highest active compound (compound 54) can be nicely mapped onto the Hypo1 model by the best fit values, which are shown in Figure 2A (1) indicating that the Hypo1 model provides reasonable pharmacophoric characteristics of the PTP-1B inhibitors for component of their activities.

Figure 1: The best hypothesis model Hypo1 produced by the HypoGen module in Catalyst 4.11 software. The best hypothesis model Hypo 1 produced by the HypoGen module in Catalyst 4.11 software. Pharmacophore features are color-coded with orange, blue and green contours representing the ring aromatic features (RA), hydrophobic feature (HY) and hydrogenbond acceptor feature (HA) respectively. Distance between pharmacophore features is reported in angstroms.
Cost analysis
In addition to generating a hypothesis, Catalyst also provides two theoretical costs (represented in bit units) to help assess the validity of the hypothesis. The first is the cost of an ideal hypothesis (fixed cost), which represents the simples fits all data perfectly. The second is the cost of the null hypothesis (null cost), which represents the highest cost of a pharmacophore with no features and which estimates activity to be the average of the activity data of the training set molecules. They represent the upper and lower bounds for the hypotheses that are generated. A generated hypothesis with a score that is substantially below that of the null hypothesis is likely to be statistically significant and bears visual inspection.
The greater the difference between the cost of the generated hypothesis and the cost of the null hypothesis, the less likely it is that the hypothesis reflects a chance correlation. A value of 40- 60 bits between them for a pharmacophore hypothesis may indicate that it has 75-90% probability of correlating the data. The total fixed cost of the run is 119.487, the cost of the null hypothesis 309.536, and the total cost of the Hypo1 is 141.995 (Table 3).
| Hypo No. | Total cost | Cost difference (Null cost-Total cost) | Error cost | RMS | Correlation (r) | Features |
|---|---|---|---|---|---|---|
| 1 | 125.257 | 62.436 | 106.062 | 1.3258 | 0.911 | HBA, HY, RA, RA |
| 2 | 125.899 | 56.907 | 106.531 | 1.3398 | 0.927 | HBA, HY, RA, RA |
| 3 | 126.049 | 52.348 | 94.041 | 1.345 | 0.939 | HBA, HBA, HY, RA |
| 4 | 126.098 | 51.151 | 106.247 | 1.331 | 0.947 | HBA, HY, RA, RA |
| 5 | 126.478 | 50.764 | 107.118 | 1.354 | 0.966 | HBA, HY, RA, RA |
| 6 | 125.850 | 50.656 | 102.352 | 1.605 | 0.949 | HBA, HY, HY, RA, RA |
| 7 | 125.777 | 50.925 | 101.813 | 1.590 | 0.957 | HBA, HBA, HY, HY |
| 8 | 124.566 | 50.946 | 101.824 | 1.590 | 0.949 | HBA, HY, HY, RA |
| 9 | 125.594 | 49.890 | 100.769 | 1.560 | 0.956 | HBA, HBA, HY, RA, RA |
| 10 | 124.663 | 49.769 | 103.641 | 1.641 | 0.945 | HBA, HY, HY, RA |
aNull cost = 309.536, fixed cost = 119.487; configuration = 15.469 and weight ~ 1.224. All cost units are in bits.
bHBA, hydrogen-bond acceptor; HY, hydrophobic feature; HBD, hydrogen-bond donor; RA, ring aromatic feature.
Table 3: Results of pharmacophore hypothesis generated using training set against PTP-1B.
Then, the cost range between Hypo1 and the fixed cost is 25.426, while that between the null hypothesis and Hypo1 is 163.797 (Table 3), which shows that Hypo1 has more than 90% probability of correlating the data. Noticeably, the total cost of Hypo1 was much closer to the fixed cost than to the null cost. Furthermore, a high correlation coefficient of 0.966 was observed with RMS value of 1.354 and the configuration cost of 14.536, demonstrating that we have successfully developed a reliable pharmacophore model with high predictivity.
Score hypothesis
To verify Hypo1’s discriminability among PTP-1B inhibitors with different order of magnitude activity, all training set compounds were classified by their activity as highly active highly active < 0.5 uM (highly active); ++, > 0.5-10 uM (moderately active); +, > 10 uM (inactive). The actual and estimated PTP-1B inhibitory activities of the 25 compounds based on Hypo1 are listed in Table 1.
The discrepancy between the actual and the estimated activity observed for the two compounds was only about one-order of magnitude, which might be an artifact of the program that uses different numbers of degrees of freedom for these compounds to mismatch the pharmacophore model. The error factor is also reported in Table 1. It shows that 20 molecules out of the 25 molecules in the training set have errors less than 10 which means that the activity prediction of these compounds falls between 10-fold greater and 1/10 of the actual activity.
These results confirm that our hypothesis is a reliable model for describing the SAR in the training set. In this study, all but one highly active compound map the hydrogen-bond acceptor (HA) feature, and one least active inhibitor do not have this feature.
Validation of the constructed pharmacophore model
The actual activities versus estimated activity of the 125 compounds in the test are shown in Table S1 in the Supporting information. A correlation coefficient of 150 generated using the test set compounds shows a good correlation of 0.951 between the actual and the estimated activities. Detailed, 7 out of 10 highly active, 33 of 55 moderately active, and 43 of 60 inactive compounds were predicted correctly. Two highly active compounds were underestimated as moderately active; five moderately active compounds were underestimated as inactive and other seven moderately active compounds were overestimated as highly active; most of inactive compounds were overestimated as moderately active.
The most active compound 77 in the test set had a fitness score of 12.05 when mapped to the Hypo 1 as seen in Figure 2A(2) and shows that all the features are being mapped accurately.

Figure 2A: Pharmacophore mapping of the most active compound on the best hypothesis model Hypo1. (1) Compound 54 from the training set. (2) Compound 77 from the test set.
The least active compound 110 in the test set had a fitness score of 8.16 when mapped to the Hypo 1 as seen in Figure 2B (2) and shows that all the features are not being mapped accurately.

Figure 2B: Pharmacophore mapping of the least active compound on the best hypothesis model Hypo1. (1) Compound 18 from the training set. (2) Compound 110 from the test set.
In conclusion, most of the compounds in the test set were predicted correctly, which mean the hypothesis is suited for screening high active compounds from the database.
Fisher’s test
To further evaluate the statistical relevance of the model, Fisher’s method was applied. With the aid of the CatScramble program, the experimental activities in the training set were scrambled randomly, and the resulting training set was used for a HypoGen run. All parameters were adopted which were used in initial HypoGen calculation. This procedure was reiterated 30 times. None of the outcome hypotheses has lower cost score than the initial hypothesis.
Finally, cross validation using the CatScramble program available in CATALYST was applied to assess the statistical confidence of Hypo1. The goal of this type of validation is to check whether there is a strong correlation between the structures and activity. CatScramble mixes up activity values of all training set compounds and creates 19 random spreadsheets (Sarma et al., 2008).
In this validation test, we select the 95% confidence level. We employed the first hypothesis (Hypo1) as 3D-search query against the NCI database using the ‘fast flexible search’ approach implemented within CATALYST. The pharmacophore captured 302 hits from a commercially available database of 10,458 compounds. The molecules identified included a broad range of templates that were structurally diverse from the starting molecule. The hits were subsequently fitted against the Hypo1 and the highest ranking 30 compounds were selected for being further investigated as potential new structures for design of novel PTP-1B inhibitors Rituparna Sarma et al., 2008.
Model validation and knowledge based screening
The purpose of the pharmacophore hypothesis generation is not just to predict the activity of the training set compounds accurately but also to verify whether the pharmacophore models are capable of predicting the activities of compounds of the test set series and classifying them correctly as active or inactive.
The best pharmacophore hypothesis was used initially to screen the PTP-1B inhibitors. All queries were performed using the Best Flexible search databases/Spreadsheet method.
Hyporefine 1 was used to screen the known high, medium and low active inhibitors of the test set. Database mining was performed in Catalyst software using the BEST flexible searching technique.
A number of parameters such as hit list (Ht), number of active percent of yields (%Y), percent ratio of actives in the hit list (%A), enrichment factor of (E), False negatives, False positives and Goodness of hit score (GH) are calculated (Table 4) while carrying out the pharmacophore model and Virtual screening of test set molecules.
| S. No | Parameter | PTP-1B |
|---|---|---|
| 1 | Total molecules in database (D) | 302 |
| 2 | Total Number of actives in database (A) | 215 |
| 3 | Total Hits (Ht) | 218 |
| 4 | Active Hits (Ha) | 201 |
| 5 | % Yield of actives [(Ha/Ht)*100] | 92.2 |
| 6 | % Ratio of actives [(Ha/A)*100] | 93.49 |
| 7 | Enrichment factor (E) [(Ha*D)/(Ht*A)] | 1.3 |
| 8 | False Negatives [A - Ha] | 14 |
| 9 | False Positives [Ht - Ha] | 17 |
| 10 | Goodness of Hit Score [a] | 0.76 |
[a]-[(Ha/4HtA)(3A + Ht) _ (1 _ ((Ht _ Ha)/(D _ A))]; GH score of 0.6–0.7 indicates a very good model.
Table 4: Statistical parameters from screening test set molecules.
The number of molecules in the database is 302. Of these, 215 are highly active, 54 are moderately active and 36 are low active compounds. While the False positives and negatives, 16 and 12 respectively, are minimal, enrichment factor of 1.33 against a maximum value of 3.0 is a very good indication on the high efficiency of the screening. Of the 215 highly active molecules, 15 were predicted as moderately active and 4 were predicted as least active. In the 54 moderately active, 6 were predicted as low active and 3 as highly active.
The model also predicted 3 of the low active molecules as moderately active and 2 more molecules from the same set as highly active. The steric and other interaction effects might have a subtle, yet crucial role on the predicted activity.
While these additional groups may not prevent in identifying many low energy conformers or add any penalty for the total cost, but could be detrimental to fit these conformers in the active site.
Thus the features of Hyporefine 1 are relatively well optimized. However, in the case of highly active molecules, there are bulky groups present which may decrease the ability of the hyporefine to select the most highly active molecules Rituparna Sarma et al., 2008.
The work presented in this study shows how chemical features of a set of compounds along with their activities ranging over several orders of magnitudes can be used to generate pharmacophore hypotheses that can successfully predict the activity. The models were capable of predicting the activities over a wide variety of scaffolds and showed distinct chemical features that may be responsible for the activity of the inhibitors.
This knowledge can be used to identify and design inhibitors with greater selectivity.
Thus, the pharmacophores generated from the PTP-1B can be used:
1. To generate Pharmacophore models as powerful search tool to be used as a 3D query to identify lead molecules from chemical databases as potential PTP-1B inhibitors.
2. To evaluate how well any newly designed compound maps on the pharmacophore before undertaking any further study including synthesis. Both these applications may help in identifying or designing compounds for further biological evaluation and optimization.
A total data set of test and training of 150 compounds of selective PTP-1B inhibitors whose chemical features along with their respective activities ranging over a wide range of magnitude is used to generate pharmacophore hypotheses to successfully and accurately predict the activity. A highly predictive pharmacophore model was generated based on 25 training set molecules, which had hydrogen-bond acceptor, hydrophobic, hydrophobic bond donor and ring aromatic as chemical features which described their activities towards PTP-1B. The validity of the model was based on 125 test set molecules, which finally showed that the model was able to accurately differentiate various classes of PTP- 1B inhibitors with a high correlation coefficient of 0.851 between experimental and predicted activity.
This validated pharmacophore model, as such can be used as a query for identification of potential inhibitors of PTP-1B while it can also be used to validate the potential of the compound to inhibit the enzyme prior to taking any step regarding the synthesis. PTB 1B enzymes have proven to be exciting and promising novel targets for the treatment of obesity and cancer. In-house build Medichem database was useful as a powerful resource to identify many PTP-1B inhibitors with highly varied activities and chemotypes. These PTP-1B inhibitors have been retrieved from the resource and some of them have been used to general a Pharmacophore model while other inhibitors have been used for virtual screening to validate the model.
The best quantitative Pharmacophore model in terms of predictive value consisted of four features like one hydrogen-bond acceptor (HA), one hydrophobic aromatic (HY), and two ring aromatic (RA) features, which is further validated by using an large set of 378 PTP-1B inhibitors and gives a r value of 0.958. The most active molecule 54 (IC50 = 0.039 uM) in the training set fits very well with this top scoring pharmacophore hypothesis. Virtual screening produced some false positives and a few false negatives. It is being noted that concurrent use or a consensus study, which readily minimizes these errors, could be an added tool for Pharmacophore model based virtual screening in order to produce reliable true posi tives and negatives. This Pharmacophore model was further used to search the NCI database consisting of structurally diversified molecules, yielded 218 molecules as hits that satisfied the 3D query. The activities of those molecules were predicted using the developed Pharmacophore model and the highly active molecules are further used to design more potent lead molecules against PTP-1B inhibitors for the treatment of various types of diabetes and obesity.
Thus, we hope that the model generated will be helpful to identify novel and potential lead molecules with improved activity against PTP-1B.
All molecular modeling works were performed on a Silicon Graphics Octane R12000 computer running Linux 6.5.12 (SGI, 1600 Amphitheatre Parkway, Mountain View, CA 94043) Catalyst 4.11 software was used to generate Pharmacophore models.
The authors thank Dr. J.A.R.P. Sarma, Senior Vice President and S.Vadivelan, Senior Scientist, GVK Biosciences Pvt. Ltd., Chennai for their valuable guidance, providing software facilities and a great chance to work there.