Journal of Antivirals & Antiretrovirals
Open Access
ISSN: 1948-5964
ISSN: 1948-5964
Review Article - (2013) Volume 0, Issue 0
It is well known that polymeric prodrug or polymer-drug conjugate is an effective and fast growing technique for improved use of drugs for therapeutic applications. Polymer conjugated drugs generally exhibit prolonged half-life, higher stability, water solubility, lower immunogenicity and antigenicity and specific targeting to tissues or cells. Polymers are used as carriers in polymeric prodrugs/macromolecular prodrugs for the delivery of drugs, proteins, targeting moieties, and imaging agents.
The polymeric pro-drug can be regarded as drug delivery systems that exhibit their therapeutic activities by means of releasing smaller therapeutic drug molecules from a polymer chain molecule for a prolonged period of time which results in enhanced pharmacokinetic behaviour by increasing the t1/2, bioavailability, and hence prolonged pharmacological action. The potential of the polymer-drug conjugates have already been proved by success of many products in the market for the treatment of different diseases.
A model for macromolecular pro-drugs was first proposed by Ringsdorf in the mid 1970s. The pro-drug shows particular properties determined by the macromolecule presence and manifested in the pharmacokinetic behaviour of the drug-polymer conjugate. This review deals with the Rational for design of polymer-drug conjugates, requirements for selection of drug candidate for polymeric prodrug, requirements for selecting polymers as candidate drug carriers, classification of polymers, design and synthesis of polymeric prodrugs, strategies to reduce steric hindrances exhibited by polymers and the bio-components, strategies to enhance the reactivity of polymer and the drug by incorporation of spacers and structure-activity relationship (SAR) of polymer-drug conjugates.
Keywords: Polymeric prodrug; Polymers; Ringsdorf’s model; Enhanced pharmacokinetic behavior; Drug-polymer conjugate
Improving the therapeutic index of drugs is a major impetus for innovation in many therapeutic areas. Conventional drug delivery systems (tablets, capsules, pills, suppositories, creams, ointments, liquids, aerosols and injectables, as drug carriers) involving a number of non-specific administrations of a drug may have contraindications such as variable drug concentrations and excessive drug presence also in healthy tissues, with toxic and side effects [1,2]. Frequent high doses are often needed because of the drug’s unfavourable physical and chemical properties, such as short biological half life, excessive or insufficient water-solubility, which restricts drug bioavailability, and also because of low drug specificity towards the affected organs [3]. To overcome these problems two main approaches are possible: the use of new therapeutic substances or a more effective use of known drugs by means of new therapeutic systems [4]. In recent years the second approach has been widely followed because of difficulties in the synthesis and purification of new active molecules and recent improvements in pharmaceutical technology [5].
Different drug delivery systems have been developed in the last few years to improve the pharmacokinetic and pharmacodynamic profile of such compounds [6]. These approaches are based on the preparation of more favourable genetic variants or on tailor-made formulations of the drug, as liposomal preparations, controlled release systems, covalent modifications of the drug by low molecular weight reagent or by polymer conjugation. The last one is a fast growing technique that already produced several molecules available in the market [7], as shown in Table 1. Also, the vast majority of clinically used drugs are low-molecular-weight compounds that exhibit a short half-life in the blood stream and a high overall clearance rate. They diffuse rapidly into healthy tissues and are distributed evenly within the body. Relatively small amounts of the drug reach the target site, and therapy is associated with side effect. A number of macromolecular delivery systems using polymers are under investigation to circumvent these limitations and improve the potential of the respective drug [8].
| S. No. | Conjugate | Indication | Marketing year | Company |
|---|---|---|---|---|
| 1 | PEG–adenosine deaminase (Adagen) | SCID syndrome | 1990 | Enzon |
| 2 | PEG–asparaginase (Oncaspar) | Acute lymphoblastic leukaemia | 1994 | Enzon |
| 3 | SMANCS (Zinostatin, Stimalamer) | Hepatocellular carcinoma | 1993 | Yamanouchi Pharmaceutical |
| 4 | Linear PEG–interferon α2b (PEG–Intron) | Hepatitis C, clinical evaluation on cancer, multiple sclerosis and HIV/AIDS | 2000 | Schering Plough/Enzon |
| 5 | Branched PEG–interferon a2a (Pegasys) | Hepatitis C | 2002 | Roche/Nektar |
| 6 | PEG–growth hormone receptor antagonist | Acromegaly | 2002 | Pfizer (Pharmacia) |
| 7 | PEG–G-CSF (Pegfilgrastim, Neulasta) | Prevention of neutropenia associated with cancer chemotherapy | 2002 | Amgen |
| 8 | Branched PEG–anti-VEGF aptamer | Age-related macular degeneration | 2004 | EyeTech Pharmaceuticals (now OSI Pharmaceutical)/Pfizer |
| 9 | PEG–anti-TNF Fab | Rheumatoid arthritis and Crohn’s disease | 2008 | UCB (formerly Celltech) |
Table 1: Polymer-drug conjugates available in market.
During the past two decades, scientists have focused their attention on developing site specific drug delivery systems and various polymers have shown promising results in this area [9]. A conjugation of a drug with a polymer forms so-called ‘polymeric prodrug’. A prodrug is a form of a drug that remains inactive during its delivery to the site of action and is activated by the specific conditions in the targeted site. In other words, a prodrug is an inactive precursor of a drug. The concept of the prodrug was first suggested by Albert [10] and Harper [11].
Polymer materials were designed and proposed as matrices or depot systems for injectable or implantable systems or devices. Prof. H. Ringsdorf developed a rational model (Figure 1) of polymeric prodrug for the first time in 1975. Prof. Ringsdorf was the first to recognise the immense potential of polymeric prodrugs, if only polymer chemists and biologists would work together in the field [12].

Figure 1: Ringsdorf’s model of polymeric prodrug.
The proposed model consists mainly of five components: the polymeric backbone, the drug, the spacer, the targeting group and the solubilising agent. The polymeric carrier can be either an inert or a biodegradable polymer. The role of spacer is to control the site and the rate of release of the active drug from the conjugate by hydrolytic or enzymatic cleavage [13,14]. The drug must be covalently bonded to the polymer and must remain attached to it until the macromolecule reaches the desired site of action. The choice of drug for use in this system is based on three criteria. First, only potent drug can be used because there is restriction on the amount of drug that can be administered. Second, the drug should have a functional group by which it can bind with the polymer backbone directly or by means of spacer molecule. Third, the drug must be sufficiently stable and should not be excreted in this conjugate form until it is released at the desired site [15].
The targeting moiety or homing device guide the entire drugpolymer conjugate to the targeted tissue. The targeting ability of the delivery system depends on the several variables including: receptor expression; ligands internalization; choice of antibody, antibody fragments or non-antibody ligands; and binding affinity of the ligand. [16].
Prolongation of drug action
The duration of action of the drug is determined by its plasma concentration which is usually measured as area under curve (AUC). The drugs which have slow renal elimination and are metabolically inactive have prolonged duration of action. The duration of action can be prolonged by linking a drug to a polymer in order to obtain a conjugate. This conjugation results in a slower renal excretion, longer blood circulation and an endocytotic cell uptake [17-19].
Controlled drug release
The polymeric prodrug formed by conjugation of drug with polymeric carrier should be stable in circulation but should also be able to release the macromolecular drug intra-cellularly or intra-tumorally for therapeutic effect. This controlled release from polymeric prodrug can only be achieved by proper selection of linkage between drug and polymeric carrier.
i) pH controlled drug release: The therapeutic effect is achieved only when the macromolecular drug from the polymeric prodrug is released intracellularly in the lysosomes or tumour tissue which are slightly acidic in comparison to the healthy tissues [20]. This relatively low pH has been exploited to design pH sensitive spacers such as N-cis-aconityl spacer [21] used to form polymeric prodrug of daunorubicin-linked aminoethyl polyacrylamide beads and poly(dlysine) and Hydrazon linkage used to form cytotoxic adriamycin immunoconjugates [22] which showed highest in vitro and in vivo anti-tumor activity.
ii) Enzymes for drug release: When the polymeric prodrug is up taken intracellularly, it enters the lysosomes which are present in normal as well as tumor tissues. In the lysosomes, the polymeric prodrug is acted upon by lysosomal enzymes such as cathepsins and metalloproteinases to release the macromolecular drug [23]. The release of cytotoxic drug with the help of these enzymes destroys the tumor tissue. Examples of such conjugation include coupling of mescaline with poly(vinylpyrrolidone-coacrylic acid) [24] in order to increase the biological half life of mescaline.
Enhanced permeability and retention effect
The polymeric prodrugs are taken up by solid tumors by pinocytosis and this passive tumor uptake increases the targeting of drug due to their characteristic feature of enhanced permeability and retention effect [25]. This effect is due to increased tumor vascular permeability and poor tissue drainage from the tumor cells which increase the duration of action and targeting of the macromolecular drug (Figure 2) [26].

Figure 2: The EPR effect.
The tumor cells contain permeability enhancing factors such as vascular endothelial growth factor (VEGF), bradykinin etc., which increase the permeability of polymeric prodrugs towards tumor tissue and also, lack of effective lymphatic drainage from the tumor tissue increases its retention [27].
Active targeting by Polymeric prodrug
i) Monoclonal antibodies: The monoclonal antibodies can be used as targeting group for coupling with the drug to increase the specific targeting of the prodrug on the tumor cells [28,29]. These antibodies bind very specifically to tumor cells and this approach has been successfully used in cancer therapy. For example a) Conjugate of plant toxins and antibodies, referred as immunotoxin is a very potent antitumor therapy b) Tumor selective monoclonal antibody is covalently attached to an enzyme which converts non toxic prodrug into potent cytotoxic drug after specific targeting at the tumor site [30]. This approach minimizes non specific toxicity.
ii) Lectins: The sugar specific receptors present on the plasma membrane are called lectins and they have been characterized mainly on hepatocytes [31]. Galactose specifically targets these lectins and this targeting seems to be an attractive approach for target specific drug delivery especially for treatment of liver diseases such as hepatitis, parasitic infections and liver metastasis. Drug delivery to macrophages (e.g. Kupffer cells) can be employed for targeted treatment of various malfunctions such as leishmaniasis, Gaucher’s syndrome etc., [32].
iii) Angiogenic vessels of tumor cells: The endothelial cells in angiogenic vessels of tumors show increased expression of cell surface proteins. These proteins include receptors for vascular endothelial growth factor (VEGF) and integrin receptors [33]. The peptides which specifically bind to these receptors can be used as targeting moiety for drug delivery such as RGD (arginine-glycine-aspartic acid) containing peptides that specifically bind with integrin receptors. The conjugation of RGD peptides and poly(ethylene glycol) (PEG) [34] showed increased efficacy of drug against breast cancer.
Immunoprotection by polymeric prodrugs
Treatment of cancer by polymeric prodrug can remarkably protect the patient’s immunity owing to a mechanism known as Fas-Fas ligand interaction [35]. Fas and Fas ligand (Fas L) are present on cancer as well as immune cells. Interaction between Fas and Fas ligand (Fas L) triggers a cascade of signals, that eventually results in apoptosis i.e. programmed cell death [36]. It has been reported that treatment with free anti-tumor drugs promotes induction of Fas ligands on cancer cells whereas their macromolecular derivatives did not increase Fas L [37]. This is an important outcome that might indicate that polymeric prodrugs are able to protect the patient’s immune system.
The delivery of biomolecules using polymeric materials has attracted considerable attention from polymer chemists, chemical engineers and pharmaceutical scientists. While designing the polymeric conjugates considerable attention has to be given on selection of proper drug and polymer candidate.
The properties of a drug candidate have already been mentioned while explaining the Ringsdorf’s model in the introduction section.
Requirements for selecting polymers as candidate drug carriers
• Availability of suitable functional groups -COOH, -OH, -SH or -NH2 for covalent coupling with drugs;
• Biocompatibility: preferably nontoxic, non immunogenic;
• Biodegradability or a molecular weight below the renal excretion limit;
• Availability: reproducibly manufactured and conveniently administered to patients;
• Water solubility: hydrophilic to ensure water solubility;
• Low polydispersity, to ensure an acceptable homogeneity of the final conjugates [38].
Classification of polymers used for bioconjugation
Some important polymers are classified below based on their origin:
Synthetic polymers: Synthetic polymers can be widely used because the properties of these molecules can be modified by varying there structures. The commonly used polymers of this class are:
1) Polyethylene glycol (PEG) is a polyether compound with many applications from industrial manufacturing to medicine. The structure of PEG is H-(O-CH2-CH2)n-OH.
Physical properties: It is liquid or low melting solid, clear to white in color, PEG is soluble in water, methanol, benzene, and dichloromethane, and is insoluble in diethyl ether and hexane, available over a wide range of molecular weights from 300 g/mol to 10,000,000 g/mol.
Advantages of PEG: PEG is particularly attractive because, PEG is used as a pharmaceutical excipient and is known to be non-toxic and non-immunogenic. PEG has a flexible, highly water-soluble chain that extends to give a hydrodynamic radius of effective range. Its high degree of hydration means the polymer chain effectively has a ‘water shell’, and this helps to mask the drug to which it is bound. PEG can be prepared with a single reactive group at one terminal end, and this aids site-specific conjugation to a drug and avoids cross linking during conjugation.
Disadvantage: In the macromolecular PEG- drug conjugate the overall drug content is poor since one PEG molecule has only two reactive groups, therefore at most only two drug molecules can be attached to a bulky PEG molecule. This results in low polymer-drug loading [39].
2) Vinyl polymers: Prepared by radical polymerization of the corresponding vinyl monomer. This co-polymerization results in formation of varied polymers with different polymer properties. Examples of this co-polymerization are molecules like:
a) N-(2-hydroxypropyl)methacrylamide (HPMA)
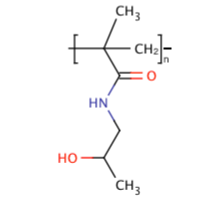
Physical properties: Molar mass: 143.18 g/mol, white odorless crystal, highly water soluble.
Advantages: Non immunogenic, non toxic, resides in blood circulation well. It is frequently used as macromolecular carriers for low molecular weight drugs (especially anti-cancer chemotherapeutic agents) to enhance therapeutic efficacy and limit side effects [40].
Poly(HPMA)-drug conjugate preferably accumulates in tumors via the passive-targeting process or the EPR effect).
It has been used as anti-tumor agent in rats after coupling with adriamycin [15,41]. This polymeric prodrug of adriamycin and HPMA has reached Phase II clinical trial for the treatment of breast, colon and lung cancer [42]
b) Poly(styrene-co-maleic acid/anhydride) (SMA)
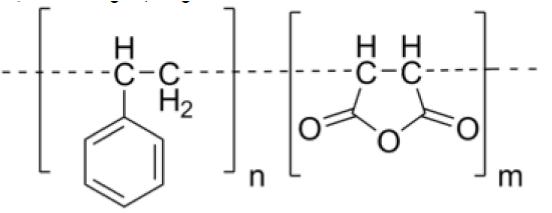
SMA or SMAnh, is a synthetic polymer that is built-up of styrene and maleic anhydride monomers.
Physical properties: Variable molecular mass, Soluble in alkaline solutions and polar organic solvents.
Advantage: Ampiphillic nature of SMA is utilized in stable micelle formation.
It forms conjugate with Neocarcinostatin (NCS) to form a polymeric prodrug ‘SMANCS’. This prodrug has been successfully marketed in Japan for the traetment of hepatocellular Carcinoma [43].
3) Divinylethermaleic anhydride/acid copolymer (DIVEMA)
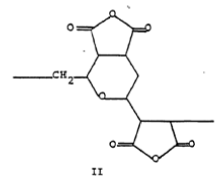
Divinyl ether (DVE) and maleic anhydride (MA) copolymerize in a 1:2 ratio with a radical catalyst. Simple acronym, DIVEMA, for divinyl ether-maleic anhydride copolymer is generally used.
The 1:2 divinyl ether-maleic anhydride cyclic alternating copolymer (DIVEMA) shows a wide variety of biological activities [44].
Advantages: It has antitumor activity; it induces the formation of interferon; it has antiviral, antibacterial, and antifungal activity; it is an anticoagulant and an anti-inflammatory agent. DIVEMA is an immunopotentiator; it increases the rate of phagocytosis, it activates macrophages selectively, and it inhibits RNA-dependent DNA polymerase.
Disadvantages: pyrogenicity, thromobocytopenia, inhibition of microsomal enzymes, sensitization to endotoxin, liver damage, organomegaly, and depression of the reticuloendothelial system.
4) Polyethylenimine (PEI) or polyaziridine is a polymer with repeating unit composed of the amine group and two carbon aliphatic CH2CH2 spacer. Linear polyethyleneimines contain all secondary amines, in contrast to branched PEIs which contain primary, secondary and tertiary amino groups. Totally branched, dendrimeric forms were also reported [45].
Properties: The linear PEIs are solids at room temperature where branched PEIs are liquids at all molecular weights. Linear polyethyleneimines are soluble in hot water, at low pH, in methanol, ethanol, or chloroform. They are insoluble in cold water, benzene, ethyl ether, and acetone. They have a melting point of 73-75°C. They can be stored at room temperature.
Advantage: Linear PEIs of mol wt 22,000 are best to overcome nuclear barrier [46] and yields the highest transfection rates [47].
Disadvantage: It has a limitation of relatively high toxicity and this could prove problematic for repeated systemic use.
Natural polymers:
1) Dextran:
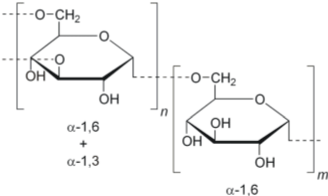
Dextran is a complex, branched glucan (polysaccharide made of many glucose molecules) composed of chains of varying lengths (from 3 to 2000 kilodaltons).
Physical properties: Molecular weight ranges from 3,000 Da to 2,000,000 Da. Dextran is neutral and water soluble. It is easily filtered.
Advantages: It is biocompatible and biodegradable. It is biologically active and posses thrombolytic activity. It is non immunogenic and non toxic.
Disadvantages: These include anaphylaxis, volume overload, pulmonary edema, cerebral edema, or platelet dysfunction. It is non immunogenic but modification of the chain with drug attachment may lead to immunogenicity and it may also create non biodegradable polymer.
2) Chitosan:
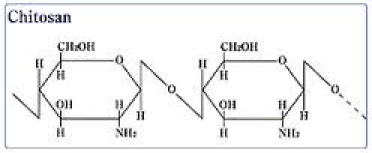
Chitosan is a linear polysaccharide composed of randomly distributed ß-(1-4)-linked D-glucosamine (deacetylated unit) and N-acetyl-D-glucosamine (acetylated unit).
Physical Properties: The molecular weight of commercially produced chitosan is between 3800 and 20,000 Daltons. Chitosan is water soluble and a bioadhesive which readily binds to negatively charged surfaces such as mucosal membranes
Advantages: Chitosan enhances the transport of polar drugs across epithelial surfaces, and is biocompatible and biodegradable. Oligomeric derivatives (3-6 kDa) are relatively nontoxic and have good gene delivery properties [48]. Chitosan is a hemostat, which helps in natural blood clotting and blocks nerve endings and hence reduces pain.
3) Proteins: Includes serum albumin which has been used extensively for preparing polymeric prodrugs with anti-viral drugs [49].
Albumin: It is playing an increasing role as a drug carrier in the clinical setting. Principally, three drug delivery technologies can be distinguished: coupling of low-molecular weight drugs to exogenous or endogenous albumin, conjugation with bioactive proteins and encapsulation of drugs into albumin nanoparticles.
Advantages: Albumin gets accumulated within solid tumors and hence is used for drug delivery and tumor targeting. It also increases the stability of attached therapeutic proteins.
4) Pullulan:
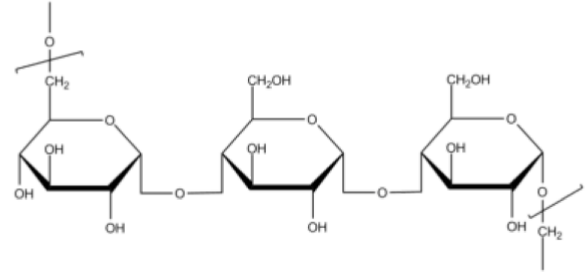
Pullulan is a polysaccharide polymer consisting of maltotriose units, also known as a-1,4-; a-1,6-glucan.
Physical properties: White powder, soluble in water. Molecular weight of pullulan ranges from thousands to 2,000,000 daltons.
Advantages: Biodegradability, low immunogenicity and polyfunctionality and fair solubility in aqueous and few organic solvents, blood compatible, non-toxic, non-mutagenic and noncarcinogenic [50].
Pseudosynthetic polymers:
1) Synthetic poly(a-amino acids): like poly(l-lysine), poly(lglutamic acid), poly((N-hydroxyalkyl)glutamines) have functional groups like amine, hydroxyl and carboxyl in their side chains that allows covalent coupling with the drug molecules [51,52]. The polymers used mostly in this class include poly(N-(2-hydroxyethyl-lglutamine)) (PHEG) which is non toxic, biocompatible and bio-degradable [53].
2) Polyglycolide or Polyglycolic acid (PGA)
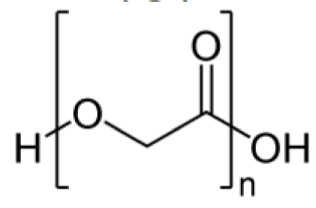
It is a thermoplastic polymer and the simplest linear, aliphatic polyester.
Physical Properties: melting point is reported to be in the range of 225-230°C. The solubility of this polyester is somewhat unique, in that its high molecular weight form is insoluble in all common organic solvents (acetone, dichloromethane, chloroform, ethyl acetate, tetrahydrofuran), while low molecular weight oligomers sufficiently differ in their physical properties to be more soluble.
Advantages: It is a biodegradable polymer that degrades in the body by simple hydrolysis of the ester backbone to non-harmful and non-toxic compounds and non Immunogenic.
Coupling methods
Coupling reactions involves conjugation of drugs or other biocomponents with polymers. In coupling reactions, the reactive functional groups on the different cross-linking or derivatizing reagents and the functional groups present on the target biomolecule are to be modified. Coupling agents mediate the conjugation of the two molecules by forming a bond with no additional spacer atom [54,55]. In case, the drug or the polymer contains more than one functional group, then the synthetic methodology to form a conjugate involves either protection or deprotection of the groups. The most commonly used strategies for coupling the components of polymeric prodrug involve use of coupling agents such as dicyclohexyl carbodiimide (DCC) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, EDCI) or use of N-hydroxysuccinimide esters which will be discussed in a more detail as a separate heading. Drugs or other biomolecules are chemically conjugated to polymers through ester, amide or disulphide bonds. The resulting bond linkage should be relatively stable to prevent drug release during its transport before the cellular localization of the drug [43,56-58].
Most of the bioconjugation strategies involve coupling reactive nucleophiles with the following order of reactivity: thiol, a-amino groups, epsilone amino group, carboxyl and hydroxyl [59,60]. The pH in the reaction and presence of steric hindrance on the coupling moiety controls this order of reactivity. Recent advancements in coupling methods involve use of homobifunctional amine or heterobifunctional coupling reagents. A number of electrophilic groups e.g. epoxides, vinylsulphones, and aziridines are capable of reacting with amines and other nucleophiles. Homobifunctional compounds such as N,N’- ethyleneiminoyl- 1-6-diaminohexane, bis-aziridin, divinyl sulphone (DVS), nitrogen mustard and bis-sulphonyl chloride can form proteinprotein linkages while heterobifunctional reagents are useful to couple amines with other functional groups. Reactive groups in proteincarboxyl functions offer alternating thiol reactions as a site for hetero bifunctional coupling with amines [15,61].
Coupling reactions involving N-hydroxysuccinimide (NHS) ester derivative: NHS shows higher reactivity at physiological pH, therefore it is used for amine coupling reactions in bioconjugation synthesis. As shown in Figure 3, the NHS ester compounds react with nucleophiles to form an acylated product with NHS as a leaving group [62]. Carboxyl groups activated with NHS esters are highly reactive with amine nucleophiles. Carboxyl groups are easily reacted with amine nucleophiles after their activation by NHS esters [63,64].

Figure 3: NHS esters compounds react with nucleophiles to release the NHS leaving group and form an acetylated product.
Coupling reactions involving coupling reagents:
i) Dicyclohexylcarbodiimide (DCC):
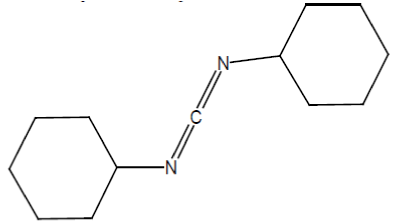
It is mainly used to couple amino acids during artificial peptide synthesis. It is highly soluble in dichloromethane, tetrahydrofuran, acetonitrile and dimethylformamide. It is insoluble in water.
A range of alcohols, including even some tertiary alcohols, can be esterified using a carboxylic acid in the presence of DCC and a catalytic amount of DMAP (Dimethyl amino pyridine) (Figure 4) [65].

Figure 4: Esterification involving DCC coupling reagent.
ii) 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, EDCI)
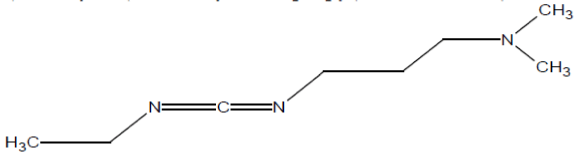
It is mainly used as a carboxyl activating agent for the coupling of primary amines to yield amide bonds. EDC is often used in combination with N-hydroxysuccinimide (NHS) or sulfo-NHS to increase coupling efficiency or create a stable amine-reactive product. EDC is also used to couple a carboxylic acid to alcohol using DMAP as a catalyst (Figure 5).

Figure 5: Formation of amide or ester bond using EDC coupling reagent.
EDC can also be used to activate phosphate groups. Biomacromolecules containing phosphate groups such as the 5’ phosphate of oligonucleotide can be conjugated to amine containing molecules by using EDC. EDC activates the phosphate to an intermediate phosphate ester. Further, in the presence of an amine carbodiimide can be conjugated to form a stable phosphoramidate bond (Figure 6) [66].

Figure 6: Phosphate oligonucleotide-polymer conjugation using EDC as couplng agent.
iii) 1-Hydroxybenzotriazole (HOBT):
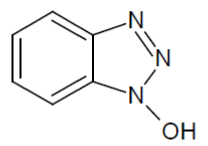
HOBt is used for the synthesis of amides from carboxylic acids aside from amino acids (Figure 7) [67,68].

Figure 7: Example of coupling reaction with HOBt.
Coupling of hydroxyl group containing polymers to alcohols and amines: Polymers containing -OH groups (e.g. PEG) can be modified to Carboxylic acid derivative by treating with acid anhydrides. PEG or mPEG can be acetylated with anhydrides (e.g. succinic anhydride) to form an ester terminating to free carboxylate groups. The resulting succinylated derivative containing free -COOH group can be further used for conjugation with drugs or proteins (Figure 8). The succinyl group incorporated in the polymer may act as spacer between the polymer and the drug which may control the site and the rate of release of the active drug from the conjugate by hydrolytic or enzymatic cleavage [69,70].

Figure 8: Conversion of PEG to Succinylated PEG containing free -COOH group and coupling of succinylated PEG to amine and alcohol using a carbodiimide as coupling agent to form (a) amide bond and (b) ester bond conjugate.
Coupling of drug and polymer through spacer: Spacers may be incorporated during bioconjugation to decrease the crowding effect and steric hindrance and control the site and the rate of release of the active drug from the conjugate. Spacers can enhance ligandprotein binding and has application in prodrug conjugates and in biotechnology. Amino acid such as glycine, alanine, and small peptides are widely used as spacers due to their chemical versatility for covalent conjugation and biodegradability [71]. The a-amino acids in peptides and proteins (excluding proline) consist of a carboxylic acid (-COOH) and an amino (-NH2) functional group attached to the same tetrahedral carbon atom which extends diversity for conjugation with hydroxyl, carboxyl or amino groups of polymer or biomolecule. Moreover, amino acid based spacers are short-chained, reactive, and biocompatible and may release the active agent from the conjugate. Di-functional amino acids such as 6-amino caproic acid (6-ACA) or 4 amino butyric acids (4 ABA) have been used as spacer arms between the polymers and the ligands for applications in biotechnology [72].
A polymeric prodrug of alkylating agent mitomycin C (MMC) with poly[N5-(2-hydroxyethyl)-L-glutamine] (PHEG) using oligopeptide spacers was designed predominantly for enzymatic degradation which released MMC with a rate dependant manner (Figure 9) [73,74].

Figure 9: A polymeric prodrug of alkylating agent mitomycin C (MMC) with poly[N5-(2-hydroxyethyl)-L-glutamine] (PHEG) using oligopeptide spacers.
Heterobifunctional coupling agents containing succinimidyl group have also been used extensively as spacers [75] (Figure 10).

Figure 10: Heterobifunctional coupling agents with spacer.
The binding ability of an antibody to its specific receptor decreases as numbers of drug molecules attached to the antibody are increased. Incorporation of a polymer chain as a spacer between the drug and the targeting polymer may overcome this binding problem and increases the potential number of drug molecules able to attach to that antibody (Figure 11) [74].

Figure 11: a) Antibody-Drug Conjugate b) Antibody-Spacer-Drug Conjugate.
Polymeric prodrugs and Steric Hindrance
The large or bulkier property of polymers causes steric hindrances for covalent conjugation with drugs as the reacting groups may not be at may not be at an appropriate distance to attract each other or they may have other atoms blocking them. Therefore some methodologies have to be adopted while conjugating drug and polymer or peptides in order to reduce the steric hindrances caused by the polymer [75-78].
The steric hindrances can be reduced by either incorporating a spacer between the polymer and the drug or by increasing the reactivity of the polymer or biomolecules. For example, A sialyl Tn (STn)- keyhole limpet hemocyanin (KLH) conjugate vaccine was synthesized using a 4-(4-N-maleimidomethyl) cyclohexane-1-carboxyl hydrazide (MMCCH) as a spacer arm to decrease the steric hindrance caused by the large STn(c) molecules during conjugation [79].
Polymeric prodrugs and targeting
In case, if the polymeric prodrug does not release the drug in the gastro intestinal tract then the prodrug are internalized mainly by endocytosis which is a much slower internalization process when compared with simple diffusion and generally requires much higher drug concentration outside the cell to produce the same cellular effect as corresponding low molecular weight drug. For example, polymeric anticancer drugs are generally less toxic when compared with free drugs yet require substantially higher concentrations inside the tumor to kill the same amount of cancer cells as low molecular weight drugs. This decrease in drug efficacy can be compensated by targeting a polymeric drug to the specific organ, tissue and/or cell [80-82]. The targeted drug acts like a ‘magic bullet’ selectively killing the villain and sparing the innocent. Drug targeting is especially important in cancer chemotherapy due to high toxicity of the drugs used.
Two approaches are mainly used for targeting polymeric prodrugs:
1) Passive targeting 2) Active targeting.
Passive targeting: Enhanced permeability and retention effect is the main approach in passive targeting and has been explained already in the section Enhanced permeability and retention effect. Passive targeting is not very efficient as the polymeric drug enter the cells by means of the concentration gradient between the intracellular and extracellular spaces [83].
Active targeting: The active targeting approach is based on the interactions between a ligand and a receptor or between a specific biological pair (e.g. avidin-biotin, antibody-antigen, lectincarbohydrate, etc.). In most cases, a targeting moiety in a polymeric drug delivery system is focused on the specific receptor or antigenoverexpressed in the plasma membrane or intracellular membrane of the targeted cells [84]. Figure 12 presents a very simplified model, illustrating interaction of the polymer prodrug with the cell and its internalization [85].

Figure 12: Interaction of the polymer prodrug with the cell. Targeting antibody in the conjugate interacts with its receptor in the cell membrane. Polymer conjugate enters the cell by receptor-mediated pinocytosis. (A) Drug is released in endosomes as a result of pH drop from 7.4 (external pH) to 5~6 (pH in endosomes) in case of the conjugates with pH-sensitive spacer. (B) Drug is released in secondary lysosome due to low pH (acid-sensitive conjugates) or due to enzymolysis (conjugates with spacers tailor-made as a substrate for any lysosomal enzyme).
Although the development of drugs for HIV infection has undergone substantial progress, numerous uncertainties persist about the best way to manage this disease. Interventions such as AIDS counselling, educational tools and antiretroviral drug therapy especially Highly Active Antiretroviral Therapy (HAART) has contributed to transforming HIV infection from a fatal to a manageable chronic infectious disease. Despite the availability of these measures, much remains to be accomplished as the number of newly reported HIV infections still remains unacceptably high [86].
In addition, the HAART presents several collateral effects, such as fatigue, nausea, sickness, diarrhoea and lipodystrophy. These symptoms contribute to a lack of treatment adhesion in the patient, resulting in a rise in the blood viral load and a decline in CD4+ T cells count, as well as an increased tolerance of anti-HIV drugs, treatment failure, increased opportunistic infections and in wasted investments [87].
Moreover, the antiretroviral therapy used for HIV/AIDS treatment is associated with dose- dependent cellular toxicity and sub-optimal pharmacokinetic properties. The major clinical toxicities associated with anti-HIV drugs are peripheral neuropathy, pancreatitis and lypidodystrophy necessitating dose reduction or discontinuance of treatment. Among their sub-optimal pharmacokinetic aspects, short plasma half-life (t1/2 ˜ 0.5-2 h) constitutes a major concern which necessitates frequent administrations to maintain therapeutic drug doses, thus increasing the incidence of unwanted side-effects, which usually compromises the adherence of the patient to the anti- HIV treatment [88]. The current oral and intravenous (I.V.) dosage forms are associated with limitation of fluctuations in plasma drug concentration with an initial high concentration that increases the risk of hematological toxicities, and subsequent low drug levels that are below the therapeutic threshold requiring twice- or thrice-daily administration. Thus, there is still a need to improve several properties of anti-HIV agents, mainly focusing in enhancing their elimination half-life and maintaining the plasma concentration of the drug within the therapeutic range to avoid dose limiting toxicities.
Therefore synthesis of a macromolecular pro-drug or polymeric prodrug of anti-retroviral drugs in which the drug is attached to a polymeric backbone can achieve a good approach to control the fluctuations of the plasma drug concentration and to improve the retention of the drug in the body by releasing the drug in a pH dependent and sustained manner. The potential advantages of this concept include minimization of drug related side effects due to controlled therapeutic blood levels instead of oscillating blood levels, improved patient compliance due to reduced frequency of dosing, reduction of the total dose of drug administered and enhanced pharmacokinetic behavior by increasing the t1/2, bioavailability, and hence prolonged pharmacological action.
ARVs such as zidovudine (AZT) and didanosine (ddI) would be ideal candidates for developing their polymeric prodrugs due to their short half-life of 1-1.5 h, necessitating frequent administration of doses, as well as their severe dose dependent side effects. ddI also undergoes acid degradation in the gastric medium. In spite of the low bioavailability from the oral route of administration in comparison to i.v route, the oral route is the most convenient for AIDS patients as they require a long-term treatment regimen. The potential of macromolecular prodrugs could be fully exploited in the anti-HIV therapy, since toxic and side effects of very long therapy could be strongly reduced, and the use of targeting portions could allow high levels of drugs in those cell groups mainly invaded by HIV. Therefore, in an attempt to improve the therapeutic potential of AZT, we have synthesized polymeric prodrugs of AZT and evaluated as a sustained drug delivery system [89]. The pro-drug was synthesized by coupling the drug to 2-hydroxyethyl methacrylate (HEMA) through a succinic spacer to get a monomeric drug conjugate which was polymerized to obtain the polymeric pro-drug. The pro-drug was subjected for in-vitro drug release study in buffers of pH 1.2 and 7.4. The hydrolytic stability of the pro-drug to pepsin was assessed in simulated gastric fluid (SGF, pH 1.2) and to a-chymotrypsin in simulated intestinal fluid (SIF, pH 7.4). The results showed that the drug release from the polymeric backbone takes place in a sustained manner over a period of 24 h, and the amount of drug released was comparatively higher at pH 7.4. At all pH conditions in the presence and absence of a-chymotrypsin, AZT was released preferentially in comparison with the succinyl derivative. The in-vivo release studies in rabbits after oral administration of AZT conjugate demonstrated a sustained release of parent drug over a period of 24 h. The pro-drug provided a significant increase in the area under the plasma concentration time curve as compared to free drug and extended the plasma half-life from 1.06 h to 8.08 h. This study suggested that, after oral administration, the drug-polymer conjugate can release AZT for prolonged periods, thus improving the pharmacokinetics of AZT and after proper dose adjustment the prodrug may be able to maintain plasma drug concentration within therapeutic window thus avoiding the dose limiting toxicities.
In an another attempt, we synthesized a polymeric prodrug of ddI for oral administration using Poly(HEMA) as drug carrier and succinic acid as spacer between drug and the polymer [90]. The prodrug was subjected for in-vitro drug release studies in buffers of pH 1.2 and 7.4 mimicking the upper and lower GIT. The results showed that the drug release from the polymeric backbone takes place in a sustained manner over a period of 24 h and the amount of drug released was comparatively higher at pH 7.4 indicating that the drug release takes place predominantly at the alkaline environment of the lower GIT rather than at the acidic environment of the upper GIT. This pH dependent sustained drug release behaviour of the prodrug may be capable of reducing the dose limiting toxicities by maintaining the plasma drug level within the therapeutic range and increasing t1/2 of ddI. Moreover, the bioavailability of the drug should be improved as the prodrug releases ddI predominantly in the alkaline environment which will reduce the degradation of ddI in the stomach acid.
Li et al. [91] synthesized a mPEG-AZT prodrug as a sustained release prodrug using a succinate diester spacer to covalently couple AZT with methoxy poly (ethylene glycol) (mPEG; MW=2000). The in vitro release was determined in hydrochloride (HCl) solution (pH 1.2) and phosphate-buffered solution (PBS; pH 6.8), which showed the release rate of AZT from the conjugate was slower than that from the free drug, suggesting its possible increased retention in gastrointestinal conditions.
Pharmacokinetic properties were evaluated experimentally by oral administration in mice. Compared to free AZT, the absorption half-life (t1/2Ka) and elimination half life (t1/2ß) of AZT released from the conjugate were both extended to 0.51 ± 0.03 h (p<0.01) and 2.94 ± 0.24 h (p<0.01), respectively. Evaluation of the in vitro anti-HIV activities showed mPEG-AZT exhibited good inhibition of HIV-1, with an EC50 value of 0.0634 mm, but it is lower than that of free AZT. These researchers concluded that the conjugate is capable of releasing the parent drug in a sustained profile, potentially providing a feasible alternative to oral administration of AZT in a clinical setting.
A sustained release polymeric prodrug of AZT for parenteral administration was synthesized by Wannachaiyasit et al. [92] using dextrin as polymeric drug carrier and succinic acid as spacer. The release in vitro of free AZT and succinoylated AZT was investigated in buffer solutions at pH 5.5 and 7.4 and in human plasma. A pharmacokinetic study in rats following intravenous administration of the conjugate showed prolonged plasma levels of AZT compared to free AZT. The use of the conjugate extended the plasma half-life of AZT from 1.3 to 19.3 h and the mean residence time from 0.4 to 23.6 h. Furthermore, the conjugate provided a significant greater area under the plasma concentration-time curve and reduced the systemic clearance of AZT. The study suggested the potential of this novel dextrin-AZT conjugate as a new intravenous preparation of AZT.
A new 5'-O-AZT prodrug was synthesized by conjugating 3'-azido- 2',3'-dideoxythymidine (AZT) with poly(oxyethylene H-phosphonate) at room temperature under Atherton-Todd reaction conditions by Troev [93]. The widely used polymer PEG was converted into poly(oxyalkylene H-phosphonate)s which has a highly reactive P-H group. The results of this study indicated strongly the formation of a potent prodrug substance that had high water solubility. The polymeric product could be hydrolyzed in a controlled manner in a medium mimicking the stomach, and demonstrated significantly decreased cytotoxicity in both the cell lines investigated.
Two kinds of chitosan-stavudine (d4T) conjugates, chitosan-Oisopropyl- 5'-O-d4T monophosphate conjugate (Cs-P-d4T) with a phosphoramide linkage and chitosan-5'-O-succinyl-d4T conjugate (Cs-S-d4T) with a succinic spacer, were synthesized by Rong et al. [94]. The anti HIV activity and cytotoxicity evaluated on MT4 cell lines revealed that the anti HIV selectivity index was in the following order Cs-P-d4T > d4T >> Cs-S-d4T. The results suggested that phosphoramide linkage may be an efficient approach to improve NRTI therapy efficacy.
The last few decades have seen the development of several antiviral drugs with therapeutic value in treating life-threatening or debilitating diseases such as those caused by HIV, hepatitis B virus, herpes viruses (such as herpes simplex virus and varicella zoster virus) and influenza virus. The field grew rapidly and many new drugs were developed but only with limited use and commercial value. The high cost of developing new drugs has also limited effective anti viral treatment. Hence, improvement of the already existing antiviral drugs sounds much more feasible.
Polymeric prodrug presents a very promising picture for improving the therapeutic index of antivirals. Various studies have been done in this area. We have tried to list a few of them. Two known antiherpetic agents, acyclovir and valacyclovir, were coupled with activated poly(ethylene glycol) [95]. In vitro drug release studies demonstrated the conjugates to be stable in buffer solutions at pH 7.4 and 5.5, while only PEG-valacyclovir was stable in a buffer solution at pH 1.2. The ability of the macromolecular conjugate to release the free drug was also evaluated in plasma, in which the most stable prodrug also proved to be PEG-valacyclovir.
The macromolecular prodrug of Acyclovir was synthesized by coupling the Drug to PEG in order to improve the problem associated with oral dosage [96]. The result showed that in enzymatic hydrolysis studies more of the product was hydrolyzed in the presence of a-chymotrypsin than pepsin. However, the release profile of the drug from the polymeric backbone was slow, which would be a way to increase the duration of activity of acyclovir.
Chemi-enzymatic synthesis of ribavirin acrylate and subsequent RAFT co-polymerization with acrylic acid was also reported by Kryger et al. [97]. It afforded a formulation of a broad spectrum antiviral drug which avoids accumulation in erythrocytes, the origin of the main side effect of ribavirin. In cultured macrophages the macromolecular prodrugs exhibited decreased toxicity while maintaining the antiinflammatory action of ribavirin
There is a wealth of literature on polymeric prodrugs which includes hundreds of papers, patents and reviews. This wealth of literature indicates the crucial role of this technique in drug delivery and therapeutics. It is well accepted that bioactive components of a macromolecular drug can be delivered more efficiently by being converted into prodrug form. Modification of biological macromolecules, anticancer drugs, peptides, and proteins in the form of polymeric prodrugs are of extreme importance in therapeutics. Polymeric prodrugs have tremendous potential for use as a sitespecific delivery system and a variety of their applications have yet to be explored. This technique of polymeric prodrug conjugation is a fascinating approach for efficient drug delivery and appears to have a bright future in therapeutics.